Consider the following pedigree given in class: 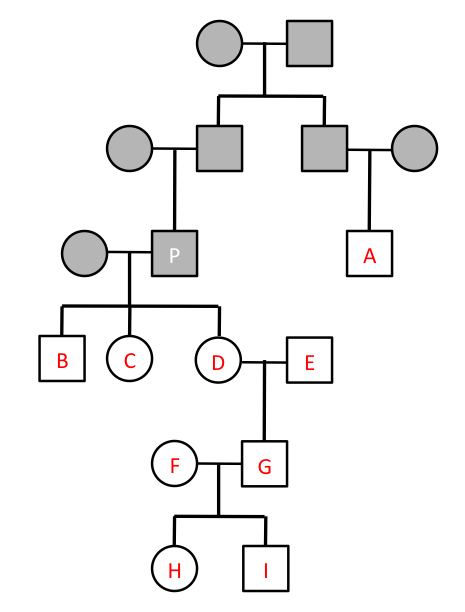
The following functions read the contents of 23andMe genotypes
def GetAlleles(filename):
""" Read the alleles in filename """
allele = dict()
with open(filename, 'rb') as tsvfile:
for row in tsvfile:
line = row[:row.find('#')].strip(' \r\n') # strip comments and line endings
if (len(line) == 0): # skip empty lines
continue
field = line.split('\t')
if (len(field) != 4): # print error for lines missing fields
print "ERROR: %s" % str(field)
continue
allele[field[0]] = field[3]
return allele
def GetCoordinates(filename):
""" Read the chromosome and positions of alleles in filename """
chrPos = dict()
with open(filename, 'rb') as tsvfile:
for row in tsvfile:
line = row[:row.find('#')].strip(' \r\n') # strip comments and line endings
if (len(line) == 0): # skip empty lines
continue
field = line.split('\t')
if (len(field) != 4): # print error for lines missing fields
print "ERROR: %s" % str(field)
continue
chrPos[field[0]] = (field[1], int(field[2]))
return chrPos
Now let's test it on an example by printing 20 random alleles
genomePosition = GetCoordinates("genomeA.txt")
allelesA = GetAlleles("genomeA.txt")
N = 0
for gId, gType in allelesA.iteritems():
chromo, pos = genomePosition[gId]
print "%15s, %2s, %10d, %2s" % (gId, chromo, pos, gType)
N += 1
if (N == 20):
break
print "alleles = ", len(allelesA)
Next well add functions to get simple allele stats.
def CountGenotypes(genotypeDict):
""" Counts the distinct genotypes in filename """
typecount = dict()
for gId, gType in genotypeDict.iteritems():
typecount[gType] = typecount.get(gType, 0) + 1
return typecount
def ClassifyTypes(typeCountDict):
""" Combine genotype counts into classes """
sHom, sHet, sHem = 0, 0, 0
iHom, iHet, iHem = 0, 0, 0
nCnt = 0
for gtype, count in typeCountDict.iteritems():
if (len(gtype) == 1):
if (gtype in "ACGT"):
sHem += count
elif (gtype in "ID"):
iHem += count
else:
nCnt += count
else:
if (gtype[0] == gtype[1]):
if (gtype[0] in "ACGT"):
sHom += count
elif (gtype[0] in "ID"):
iHom += count
else:
nCnt += count
else:
if ((gtype[0] in "ACGT") and (gtype[1] in "ACGT")):
sHet += count
elif ((gtype[0] in "ID") and (gtype[1] in "ID")):
iHet += count
else:
nCnt += count
return (sHom, sHet, sHem, iHom, iHet, iHem, nCnt)
Let's test them too.
aTypes = CountGenotypes(allelesA)
print "Allele types =", aTypes
aClass = ClassifyTypes(aTypes)
print
print "Homozygous SNP: %d" % aClass[0]
print "Heterozygous SNP: %d" % aClass[1]
print "Hemizygous SNP: %d" % aClass[2]
print "Homozygous indel: %d" % aClass[3]
print "Heterozygous indel: %d" % aClass[4]
print "Hemizygous indel: %d" % aClass[5]
print "No-calls: %d" % aClass[6]
Consider the following pedigree given in class:
allelesD = GetAlleles("genomeD.txt")
allelesE = GetAlleles("genomeE.txt")
allelesG = GetAlleles("genomeG.txt")
FixTotal, FixError = 0, 0
HetTotal, HetError = 0, 0
for gId in allelesG.iterkeys():
momsAllele = allelesD[gId]
popsAllele = allelesE[gId]
kidsAllele = allelesG[gId]
if (momsAllele == '--') or (popsAllele == '--') or (kidsAllele == '--'):
continue
if (len(kidsAllele) == 2):
if ((momsAllele[0] == momsAllele[-1]) and (popsAllele[0] == popsAllele[-1])):
if (momsAllele[0] == popsAllele[0]):
FixTotal += 1
if (kidsAllele[0] != kidsAllele[-1]):
FixError += 1
elif (kidsAllele[0] != momsAllele[0]):
print momsAllele, popsAllele, kidsAllele
FixError += 1
else:
HetTotal += 1
if (kidsAllele[0] == kidsAllele[-1]):
HetError += 1
elif ((kidsAllele[0] not in momsAllele+popsAllele) or (kidsAllele[-1] not in momsAllele+popsAllele)):
print momsAllele, popsAllele, kidsAllele
HetError += 1
print "Fixed Alleles = %d, Trio Errors = %d, Error %% = %2.4f" % (FixTotal, FixError, (100.0*FixError)/FixTotal)
print "Opposite Alleles = %d, Trio Errors = %d, Error %% = %2.4f" % (HetTotal, HetError, (100.0*HetError)/HetTotal)
HetHetHetCount = 0
for gId in allelesG.iterkeys():
momsAllele = allelesD[gId]
if (momsAllele[0] == momsAllele[-1]):
continue
popsAllele = allelesE[gId]
if (popsAllele[0] == popsAllele[-1]):
continue
kidsAllele = allelesG[gId]
if (kidsAllele[0] != kidsAllele[-1]):
HetHetHetCount += 1
print HetHetHetCount, (100.0*HetHetHetCount)/len(allelesG)