COMP 555 – BioAlgorithms – Spring 2017
Lecture 2: Searching for patterns in data¶
|

|
|
| From Daniel Becker's VisualDNA project | ||
From Chapter 1 of "Bioinformatics Algorithms: An Active Learning Approach," Compeau & Pevzner
1
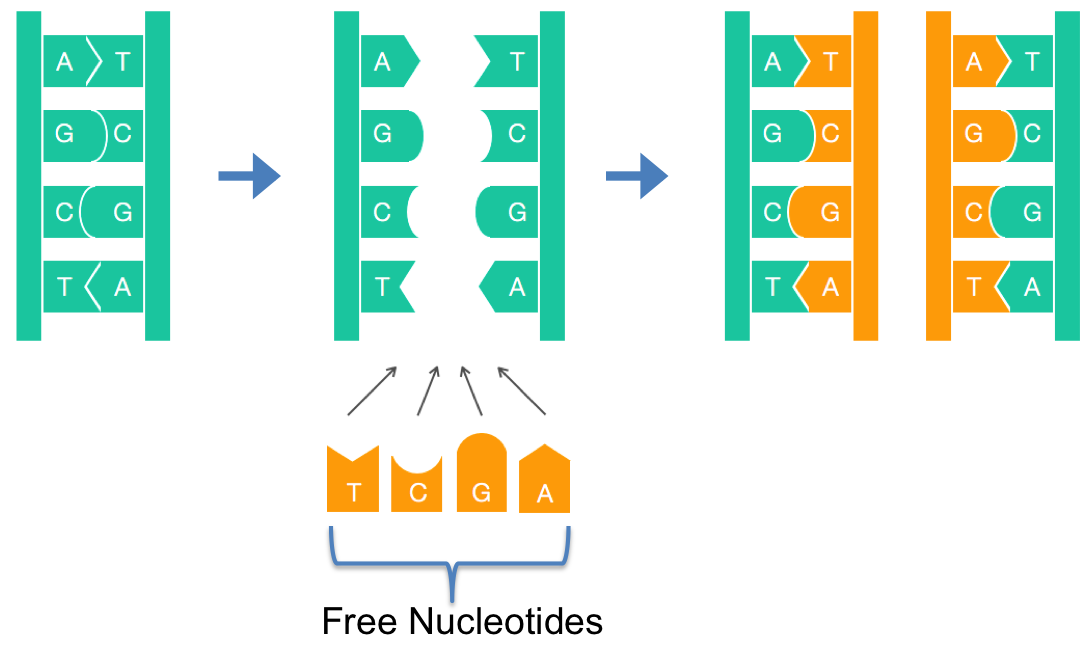
Life ≡ Reproduction ≡ Replicating a Genome
One of the most incredible things about DNA is that it provides instructions for replicating itself. Today, rather than looking for those instructions we consider how the process initiates.
2
Where Does Replication Begin?¶
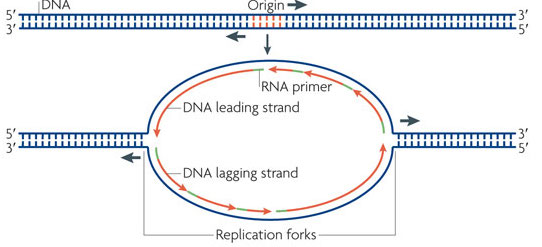
The DNA replication process begins reliably at a regions of the genome called the origins of replication or oriC. Today we investigate how these regions are identified?
3
Let's Start with Bacterial Genomes¶
In order to simplify our problem, we first consider Bacterial DNA.
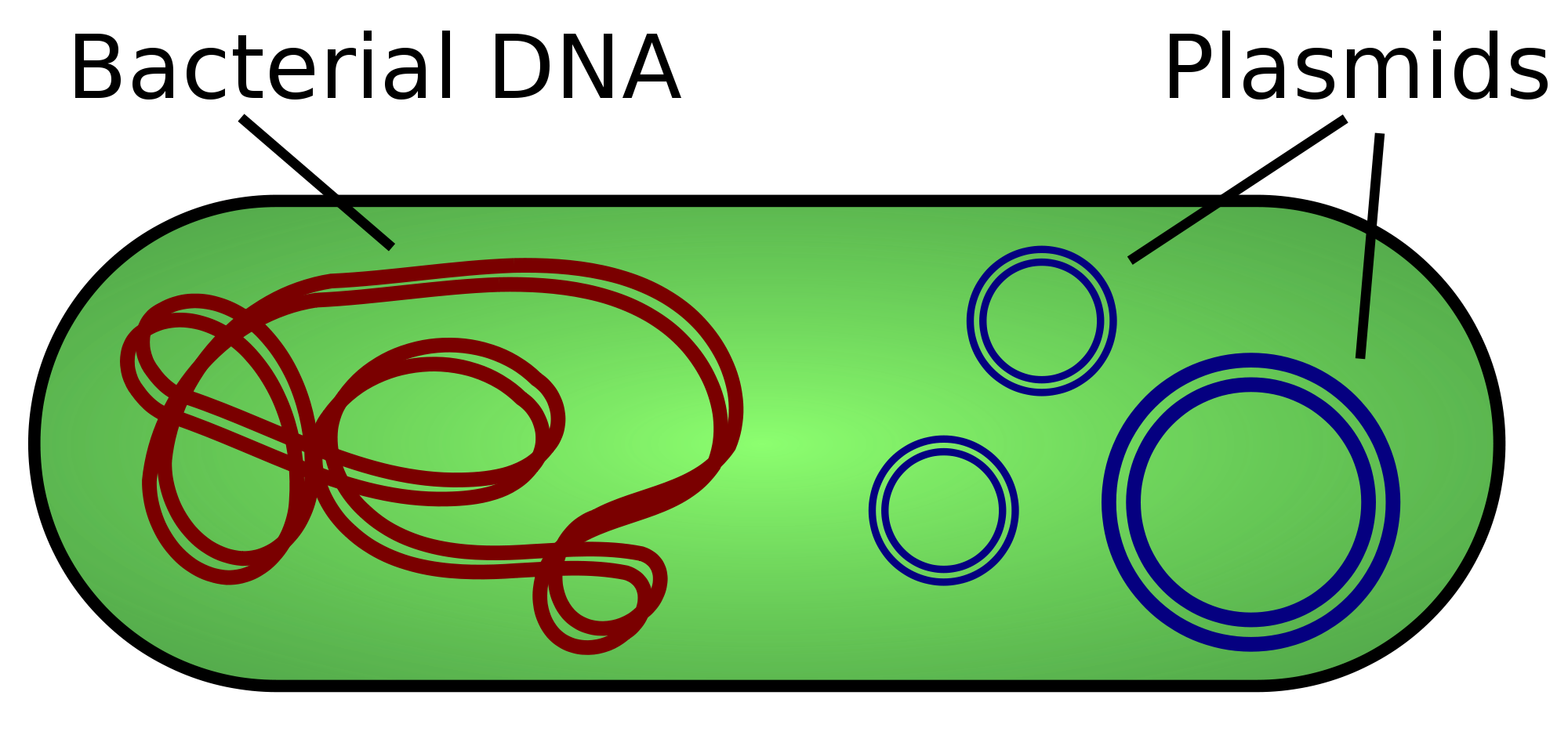
Characteristics of Bacterial DNA
- A Circular primary chromosome
- Independent, and generally smaller, circular plasmids
- Simple highly conserved mechanism
- Replication is constant (i.e. no cell cycle)
4
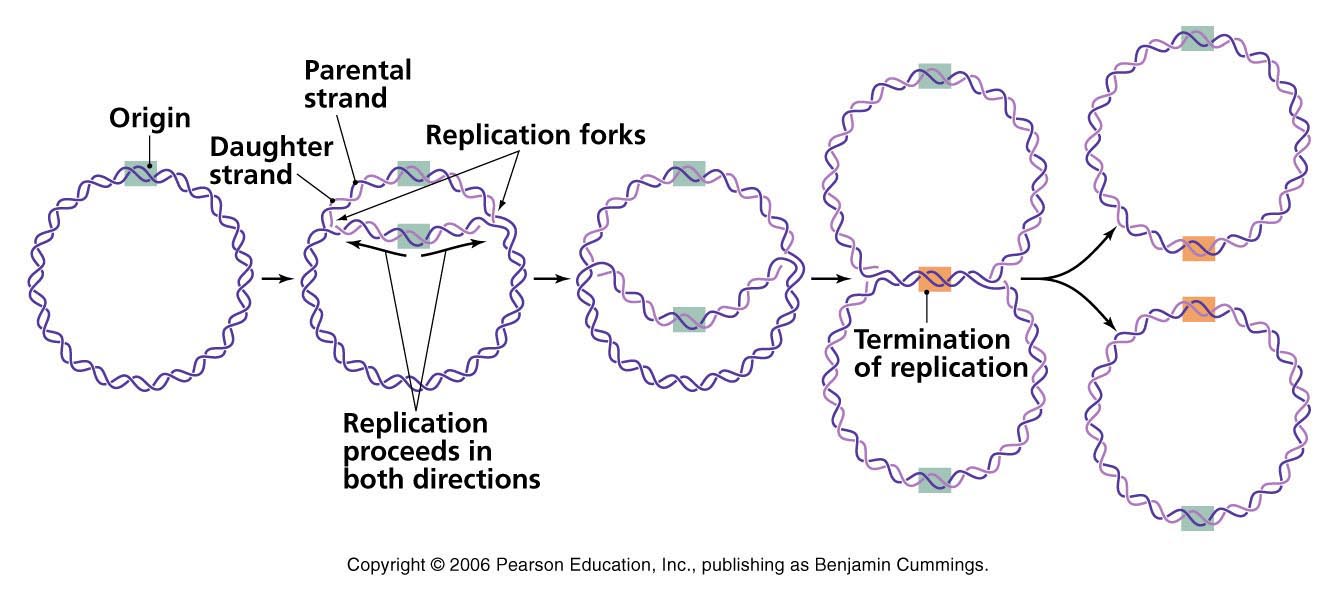
The oriC finding Problem
Biology Approach Advantage: You can start immediately
Advantage: You can start immediatelyDisadvantage: It can take a long time |
Computer Science Approach Advantage: It can be fast, and general
Advantage: It can be fast, and generalDisadvantage: Problem is not adequately specified |
6
Let's look at an example oriC¶
atcaatgatcaacgtaagcttctaagcatgatcaaggtgctcacacagtttatccacaacctgagtggatgacatcaagataggtcgttg
tatctccttcctctcgtactctcatgaccacggaaagatgatcaagagaggatgatttcttggccatatcgcaatgaatacttgtgactt
gtgcttccaattgacatcttcagcgccatattgcgctggccaaggtgacggagcgggattacgaaagcatgatcatggctgttgttctgt
ttatcttgttttgactgagacttgttaggatagacggtttttcatcactgactagccaaagccttactctgcctgacatcgaccgtaaat
tgataatgaatttacatgcttccgcgacgatttacctcttgatcatcgatccgattgaagatcttcaattgttaattctcttgcctcgac
tcatagccatgatgagctcttgatcatgtttccttaaccctctattttttacggaagaatgatcaagctgctgctcttgatcatcgtttc
Is there a pattern which might help us to develop an algorithm?
Vibrio Cholerae
Aquatic organism that causes Cholera
An abundant marine and freshwater bacterium that causes *Cholera*. Vibrio can affect shellfish, finfish, and other marine animals and a number of species are pathogenic for humans. ***Vibrio cholerae*** colonizes the mucosal surface of the small intestines of humans where it causes, a severe and sudden onset diarrheal disease.
One famous outbreak was traced to a contaminated well in London in 1854 by John Snow. Epidemics, which can occur with extreme rapidity, are often associated with conditions of poor sanitation. The disease is highly lethal if untreated. Millions have died over the centuries incuding seven major pandemics between 1817 and today. Six were attributed to the classical biotype, while the 7th, which started in 1961, is associated with this *El Tor* biotype.
7
An Aside: Accessing Sequence Data?¶
Genomes are archived as FASTA files, which are text files. Lines beginning with '>' are sequence headers. They are followed by lines of nucleotide sequences. Here's what one looks like:
!head data/VibrioCholerae.fa
!wc data/VibrioCholerae.fa
!grep ^\> data/VibrioCholerae.fa
8
A Python function to parse FASTA files¶
def loadFasta(filename):
""" Parses a classically formatted and possibly
compressed FASTA file into a list of headers
and fragment sequences for each sequence contained"""
if (filename.endswith(".gz")):
fp = gzip.open(filename, 'rb')
else:
fp = open(filename, 'rb')
# split at headers
data = fp.read().split('>')
fp.close()
# ignore whatever appears before the 1st header
data.pop(0)
headers = []
sequences = []
for sequence in data:
lines = sequence.split('\n')
headers.append(lines.pop(0))
# add an extra "+" to make string "1-referenced"
sequences.append('+' + ''.join(lines))
return (headers, sequences)
9
Example Usage¶
header, seq = loadFasta("data/VibrioCholerae.fa")
for i in xrange(len(header)):
print header[i]
print len(seq[i])-1, "bases", seq[i][:30], "...", seq[i][-30:]
print
genome = seq[0]
print "oriC:"
OriCStart = 151887
oriC = genome[OriCStart:OriCStart+540]
for i in xrange(9):
print " %s" % oriC[60*i:60*(i+1)].lower()
Outputs the header, length, the 1st 30 characters, and last 30 characters of each sequence in the file
- Note the addition of a "+" as first character
- Why might their be multiple sequences in a file?
Then it outputs a subsequence on the first sequence
10
Looking for Interesting Patterns¶
• Still not sure what "interesting" means yet
• So let's consider every pattern of a given length, k
A new well-specified problem: Find the frequency of all subsequences of length k, k-mers
atcaatgatcaacgtaagcttctaagcatgatcaaggtgctcacacagtttatccacaac
atca caacg ttctaa atcaagg acacagtt
tcaa aacgt tctaag tcaaggt cacagttt
caat acgta ctaagc caaggtg acagttta
aatg cgtaa taagca aaggtgc cagtttat
atga gtaag aagcat aggtgct agtttatc
tgat taagc agcatg ggtgctc gtttatcc
4-mers 5-mers 6-mers 7-mers 8-mers
• Let's count the occurence of every k-mer in a sequence, given a value for k. </div>
11
Code for counting k-mers¶
In a string of length N, there are N-k+1, substrings of length k
def kmerFreq(k, sequence):
""" returns the count of all k-mers in sequence as a dictionary"""
kmerCount = {}
for i in xrange(len(sequence)-k+1):
kmer = sequence[i:i+k]
kmerCount[kmer] = kmerCount.get(kmer,0)+1
return kmerCount
print kmerFreq(3, "TAGACAT")
print kmerFreq(3, "mississippi")
12
An exhaustive scan for patterns¶
- Is there some obvious pattern?
- Let's consider a range of "K" values
def mostFreqKmer(start, end, sequence):
for k in xrange(start,end):
kmerCounts = kmerFreq(k,sequence).items()
kmerCounts = sorted(kmerCounts,reverse=True,key=lambda tup: tup[1])
mostFreq = kmerCounts[0:5]
print k, mostFreq
mostFreqKmer(1,10,oriC)
13
Examine the result¶
Are two 5-mers repeated 8 times interesting? Surprizing? How about four 9-mers repeated 3 times?
14
k-mer Likelihoods¶
So we expect a specific 5-mer once per 1024 bases, so having 8 in 535 (540 - 5) bases is more than expected. We also expect a specific 9-mer once per 262,144 bases, so having 3 in 531 (540 - 9) is much more than expected.
Moreover, is their any relationship between the 9-mers ATGATCAAG and CTTGATCAT?
atcaatgatcaacgtaagcttctaagcATGATCAAGgtgctcacacagtttatccacaac
ctgagtggatgacatcaagataggtcgttgtatctccttcctctcgtactctcatgacca
cggaaagATGATCAAGagaggatgatttcttggccatatcgcaatgaatacttgtgactt
gtgcttccaattgacatcttcagcgccatattgcgctggccaaggtgacggagcgggatt
acgaaagcatgatcatggctgttgttctgtttatcttgttttgactgagacttgttagga
tagacggtttttcatcactgactagccaaagccttactctgcctgacatcgaccgtaaat
tgataatgaatttacatgcttccgcgacgatttacctCTTGATCATcgatccgattgaag
atcttcaattgttaattctcttgcctcgactcatagccatgatgagctCTTGATCATgtt
tccttaaccctctattttttacggaagaATGATCAAGctgctgctCTTGATCATcgtttc
15
Any Biological Insights to Guide us?¶
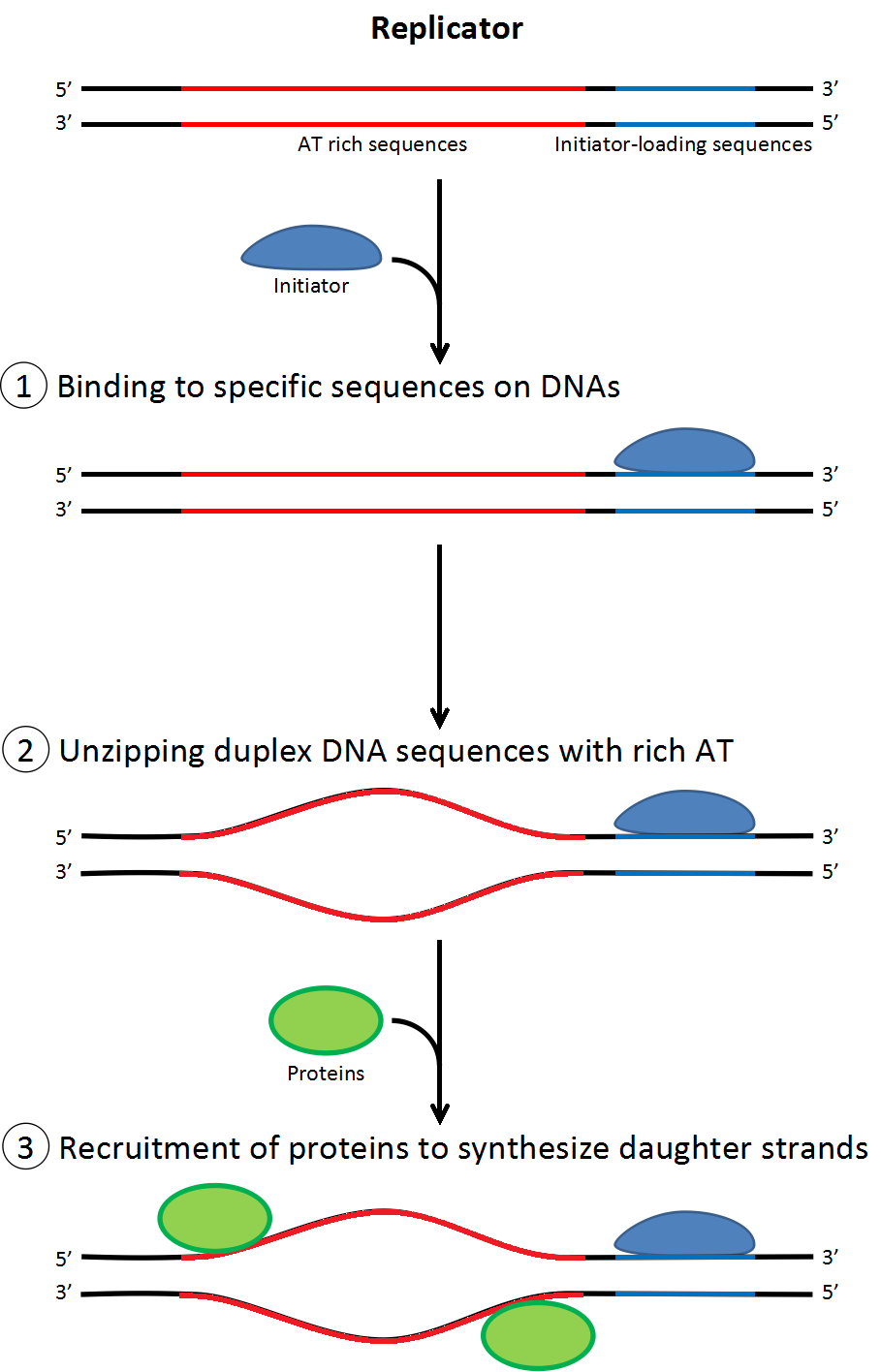
-
• Replication is performed by a DNA polymerase, and the initiation of replication is mediated by a protein called DnaA.
• DnaA binds to short (≈ 9 nucleotides long) segments within the replication origin known as a DnaA box (≈ 500 bases).
• A DnaA box is a signal telling DnaA to “bind here!”
• DnaA can bind to either strand. Thus, both the DnaA box and its reverse-complement are equal targets.
• DnaA wants to see multiple DnaA boxes.
• Sequences used by DnaA tend to be "AT-rich" (rich in adenine and thymine bases), because A-T base pairs have two hydrogen bonds (rather than the three formed in a C-G pair) which are easier to unzip. (Recall A-T was the second most common dimer with 54 after T-T with 55)
• Once the origin has been located, these initiators recruit other proteins and form the pre-replication complex, which unzips the double-stranded DNA. </ul> </div>
16
Do these Patterns Generalize?¶
aactctatacctcctttttgtcgaatttgtgtgatttatagagaaaatcttattaactgaaactaaaatggtaggtttggtggtaggttt
tgtgtacattttgtagtatctgatttttaattacataccgtatattgtattaaattgacgaacaattgcatggaattgaatatatgcaaa
acaaacctaccaccaaactctgtattgaccattttaggacaacttcagggtggtaggtttctgaagctctcatcaatagactattttagt
ctttacaaacaatattaccgttcagattcaagattctacaacgctgttttaatgggcgttgcagaaaacttaccacctaaaatccagtat
ccaagccgatttcagagaaacctaccacttacctaccacttacctaccacccgggtggtaagttgcagacattattaaaaacctcatcag
aagcttgttcaaaaatttcaatactcgaaacctaccacctgcgtcccctattatttactactactaataatagcagtataattgatctga
aaagaggtggtaaaaaa
The most frequent 9-mers are: [(ACCTACCAC,5), (GGTAGGTTT,3), (CCTACCACC,3), (AACCTACCA,3), (TGGTAGGTT,3), (AAACCTACC,3)]
There is no occurence of the patterns ATGATCAAG and CTTGATCAT
Thus, it appears that different genomes have different DnaA box patterns. Let's go back to the drawing board. By the way, the DnaA box pattern of Thermotoga petrophila is:
CCTACCACC
|||||||||
GGATGGTGG
17
A New Strategy¶
- Our previous strategy was to find frequent words in oriC region as candidate DnaA boxes, as if
replication origin → frequent words
- Suppose that we reverse our approach, we use clumps of frequent words to infer the replication origin, testing if
nearby frequent words → replication origin
- We can apply this approach to find candidate DnaA boxes.
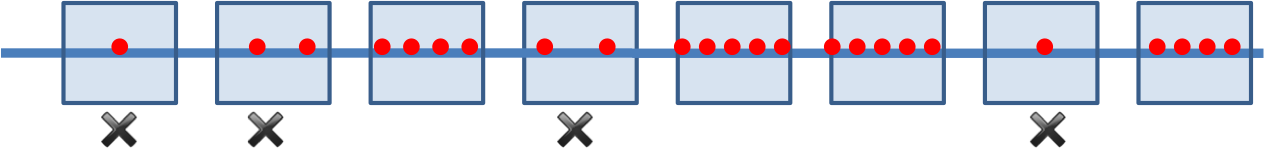
18
What is a Clump?¶
- Formal Definition:
- A k-mer forms an (L, t)-clump inside Genome if there is a short (length L) interval of Genome in which it appears many (at least t) times.
- Clump Finding Problem:
- Find patterns that form clumps within a string.
-
**Input:** A string and integers *k* (length of a pattern), *L* (window length),
and *t* (number of patterns in a clump).
**Output:** All k-mers forming (L,t) clumps in the string
19
K-mer counter that tracks positions¶
def kmerPositions(k, sequence):
""" returns the position of all k-mers in sequence as a dictionary"""
kmerPosition = {}
for i in xrange(1,len(sequence)-k+1):
kmer = sequence[i:i+k]
kmerPosition[kmer] = kmerPosition.get(kmer,[])+[i]
# combine kmers and their reverse complements
pairPosition = {}
for kmer in kmerPosition.iterkeys():
krev = ''.join([{'A':'T','C':'G','G':'C','T':'A'}[base] for base in reversed(kmer)]) # one-liner
if (kmer < krev):
if (krev in kmerPosition):
pairPosition[kmer] = kmerPosition[kmer] + kmerPosition[krev]
else:
pairPosition[kmer] = kmerPosition[kmer]
elif (kmer == krev):
pairPosition[kmer] = kmerPosition[kmer]
return pairPosition
20
Lets play a little with that one-liner.¶
mySeq = "GAGACAT"
print ''.join([{'A':'T','C':'G','G':'C','T':'A'}[base] for base in reversed(mySeq)])
print ','.join([{'A':'T','C':'G','G':'C','T':'A'}[base] for base in reversed(mySeq)])
print ' and, '.join([{'A':'T','C':'G','G':'C','T':'A'}[base] for base in reversed(mySeq)])
21
More on List Comprehensions¶
The argument of the join method is a list construction shorthand called a list comprehension. It is basically a recipe for constructing a list. Here are some simple examples.
mySeq = "GAGACAT"
print [base for base in mySeq]
print [base for base in reversed(mySeq)]
print [base for base in reversed(mySeq) if base != 'A']
22
Back to Finding Clumps¶
def findClumps(string, k, L, t):
clumps = []
kmers = kmerPositions(k, string)
for kmer, posList in kmers.iteritems():
i = 0
while (i < len(posList)-t-1):
foundSoFar = 1
for j in xrange(i+1, len(posList)):
if (((posList[j]+k) - posList[i]) > L):
break
foundSoFar += 1
if (foundSoFar >= t):
clumps.append((kmer, foundSoFar))
i = j
return clumps
23
Now let's try it¶
clumpList = findClumps(genome, 9, 500, 6)
print len(clumpList)
print [clumpList[i] for i in xrange(min(20,len(clumpList)))]
Wow, that's a lot more than expected. I guess that means that genomes are not that random at all.

24
Let's view things differently¶
# Lets get the positions of all k-mers again
kmers = kmerPositions(9, genome)
top10 = ['ATGATCAAG'] + [kmer for kmer, clumpSize in sorted(clumpList,reverse=True,key=lambda tup: tup[1])[0:9]]
print top10
%matplotlib inline
import matplotlib
import matplotlib.pyplot as plt
plt.figure(num=None, figsize=(12, 6), dpi=100, facecolor='w', edgecolor='k')
plt.plot([OriCStart, OriCStart], [0,10], 'k--')
for n, kmer in enumerate(top10):
positions = kmers[kmer]
plt.text(1120000, n+0.4, kmer, fontsize=8)
plt.plot(positions, [n + 0.5 for i in xrange(len(positions))], 'o', markersize=4.0)
limit = plt.xlim((0,1250000))

25
Summary¶

Things have not gone as planned
- We still don't have a working algorithm for finding OriC
- We tried searching for patterns in a known OriC region, but the patterns we found did not generalize to other genomes.
- We tried to find clumps of repeated k-mers, but that led to too many hypotheses to follow up on
But we won't give up
- Let's see next time if there are any more biological insights that we might leverage
26