Advanced Sequence Alignment¶

- Midterm on Wednesday
- Covers up to and including Lecture 11
- Online can be downloaded at the start of class, same Jupyter Nookbook format as Problem Sets
- Open Computer, Open Notes
- You can add extra cells for scratch work, but only the indicated answer cells will be graded
- Mix of short answer, multiple choice, and writing code fragments
- Hard deadline for submission! Bank versions as the time limit approaches.
1
Recall Local Alignment¶
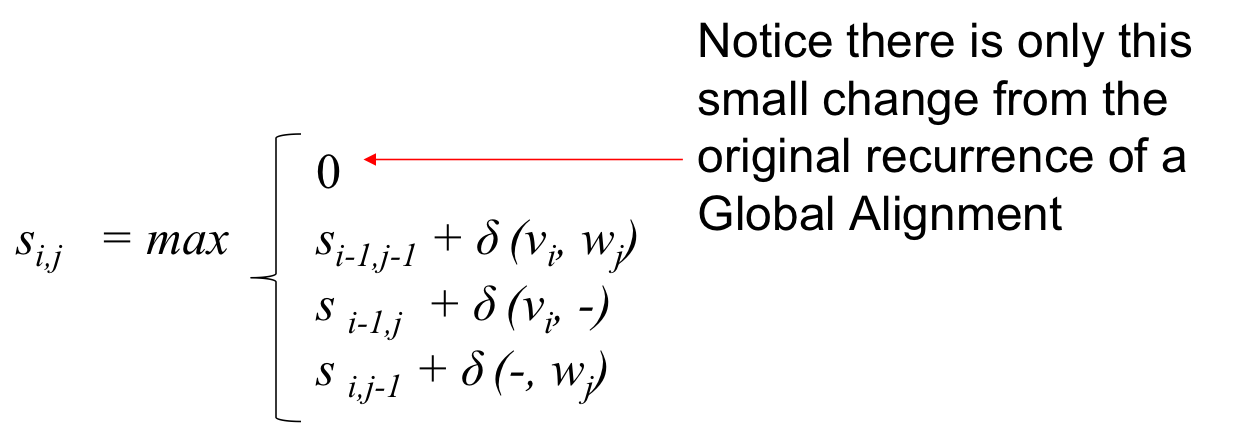
- The zero is our free ride that allows the node to restart with a score of 0 at any point
- What does this imply?
- After solving for the entire score matrix, we then search for si,j with the highest score, this is $(i_2,j_2)$
- We follow our back tracking matrix until we reach a score of 0, whose coordinate becomes $(i_1,j_1)$
2





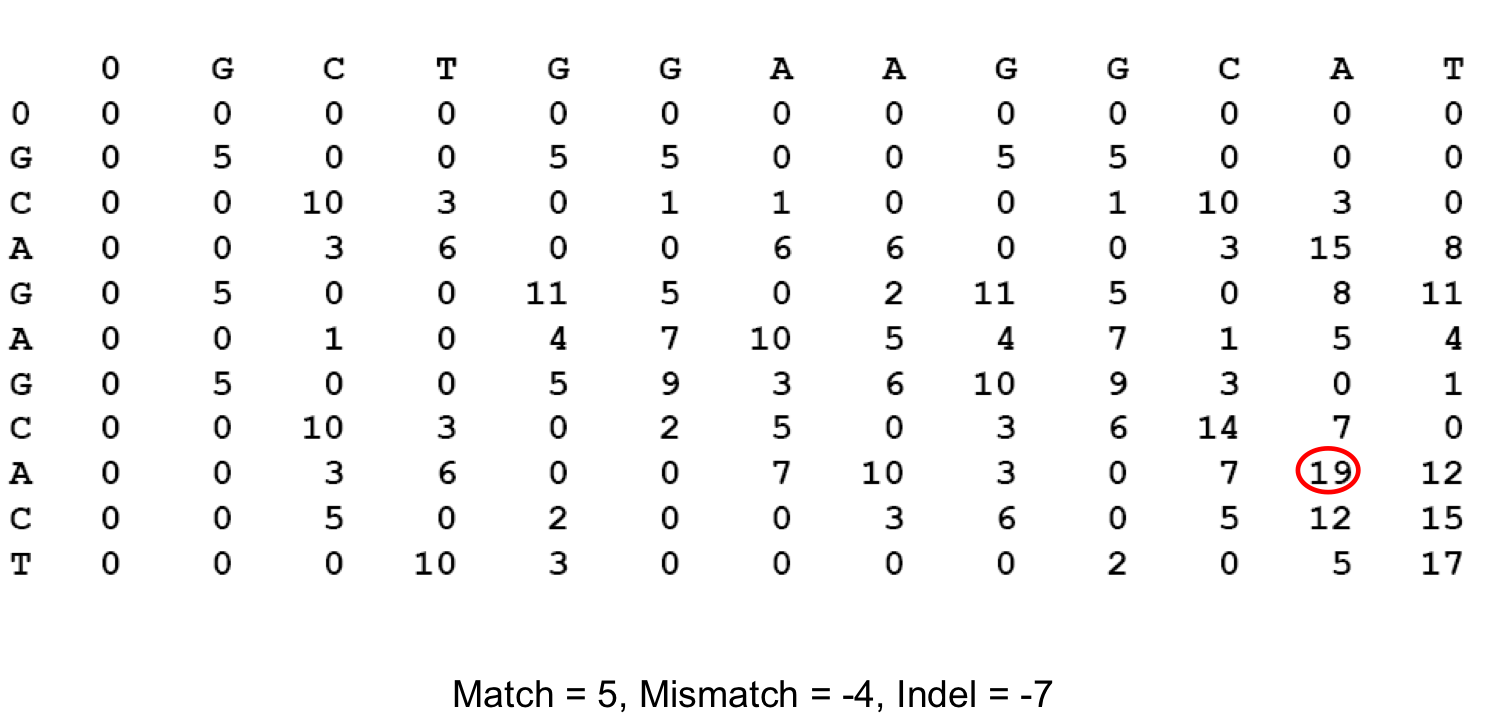
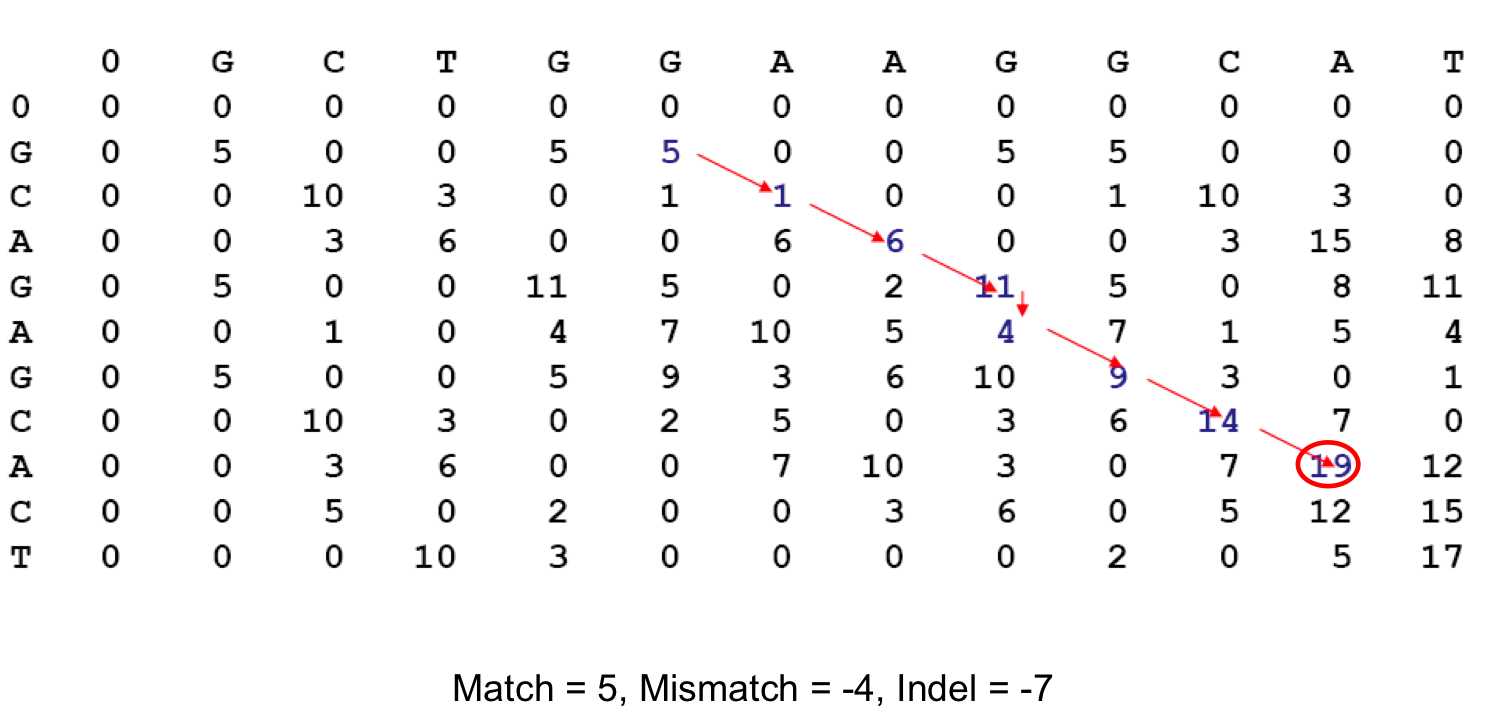
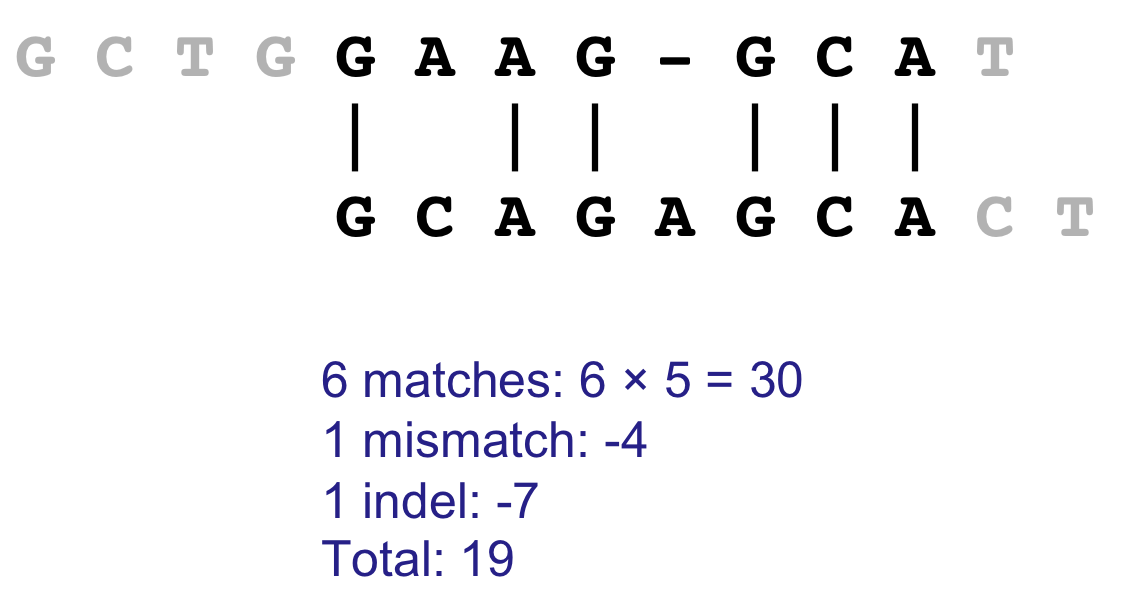
Scoring Indels: Naive Approach¶
A fixed penalty σ is given to every indel:
- -σ for 1 indel,
- -2σ for 2 consecutive indels
- -3σ for 3 consecutive indels, etc.
Can be too severe penalty for a series of 100 consecutive indels
- large insertions or deletions might result from a single event
5
Affine Gap Penalties¶
- In nature, a series of k indels often come as a single event rather than a series of k single nucleotide events:
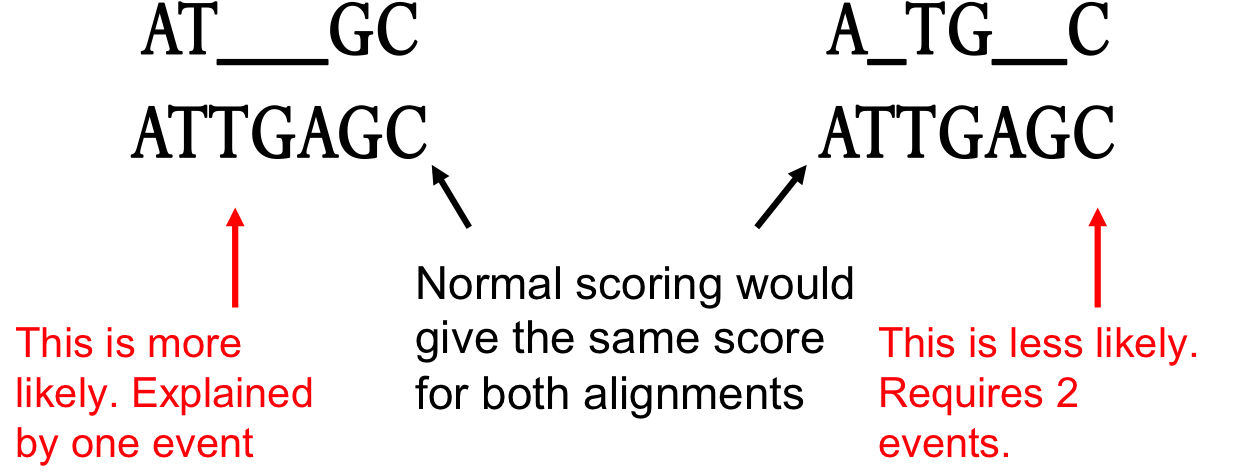
6
Accounting for Gaps¶
Gaps- contiguous sequence of indels in one of the rows
Modify the scoring for a gap of length x to be:
-(ρ + σx)where ρ+σ > 0 is the penalty for introducing a gap:
gap opening penalty
and σ is the cost of extending it further (ρ+σ >>σ):
gap extension penalty
because you do not want to add too much of a penalty for further extending the gap, once it is opened.
7
Affine Gap Penalties¶
- Gap penalties:
- -ρ - σ when there is 1 indel
- -ρ - 2σ when there are 2 indels
- -ρ - 3σ when there are 3 indels, etc.
- -ρ - x·σ (-gap opening - x gap extensions)
- Somehow reduced penalties (as compared to naïve scoring) are given to runs of horizontal and vertical edges
8
Adding Affine Gap Penalties to our Graph¶
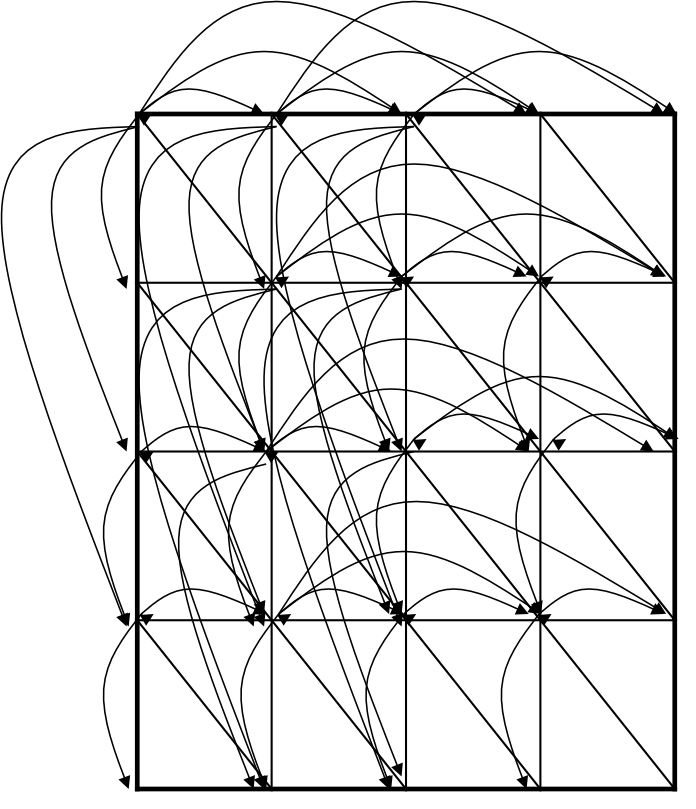
To reflect affine gap penalties we have to add “long” horizontal and vertical edges to the edit graph.
Each such edge of length x should have weight -ρ - x·σ
There are many such edges!
Adding them to the graph increases the running time of the alignment algorithm by a factor of n (where n is the number of vertices)
So the complexity increases from $O(n^2)$ to $O(n^3)$
9

A 3-level Manhattan Grid¶
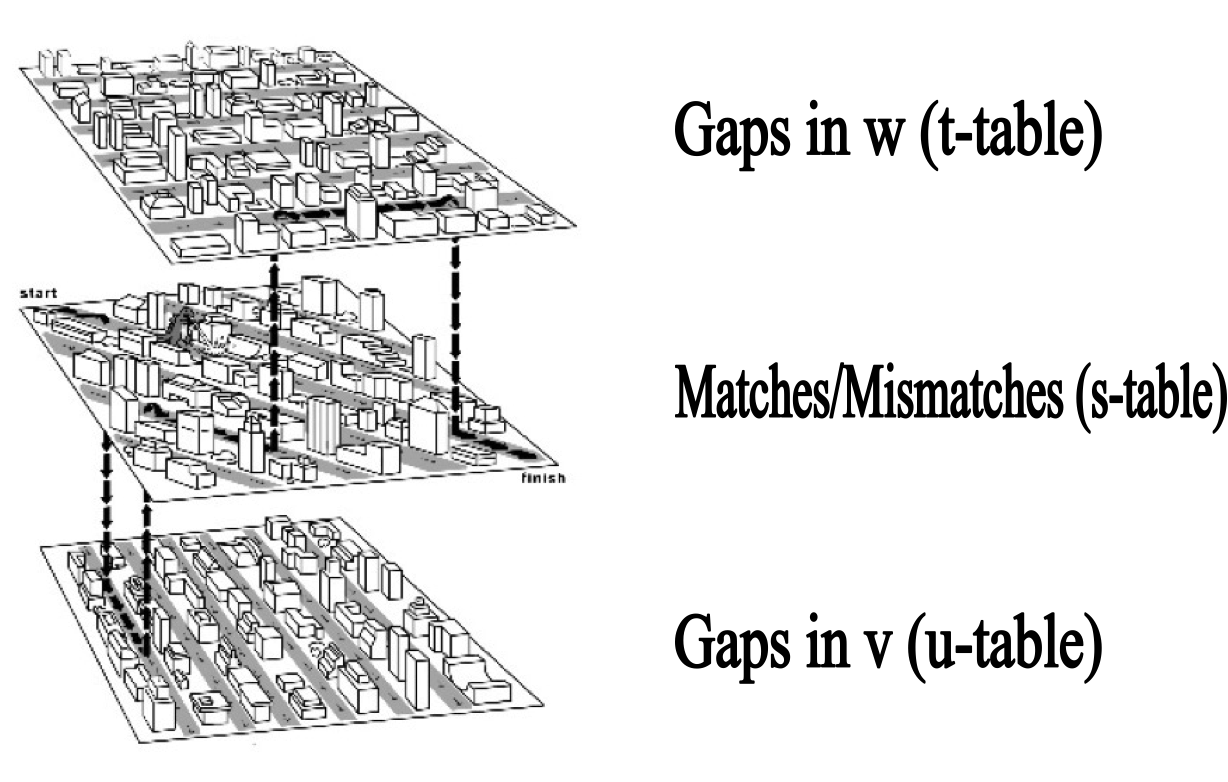
- The three recurrences for the scoring algorithm creates a 3-layered graph.
- The top level creates/extends gaps in the sequence w.
- The bottom level creates/extends gaps in sequence v.
- The middle level extends matches and mismatches.
11
Switching between 3 Layers¶
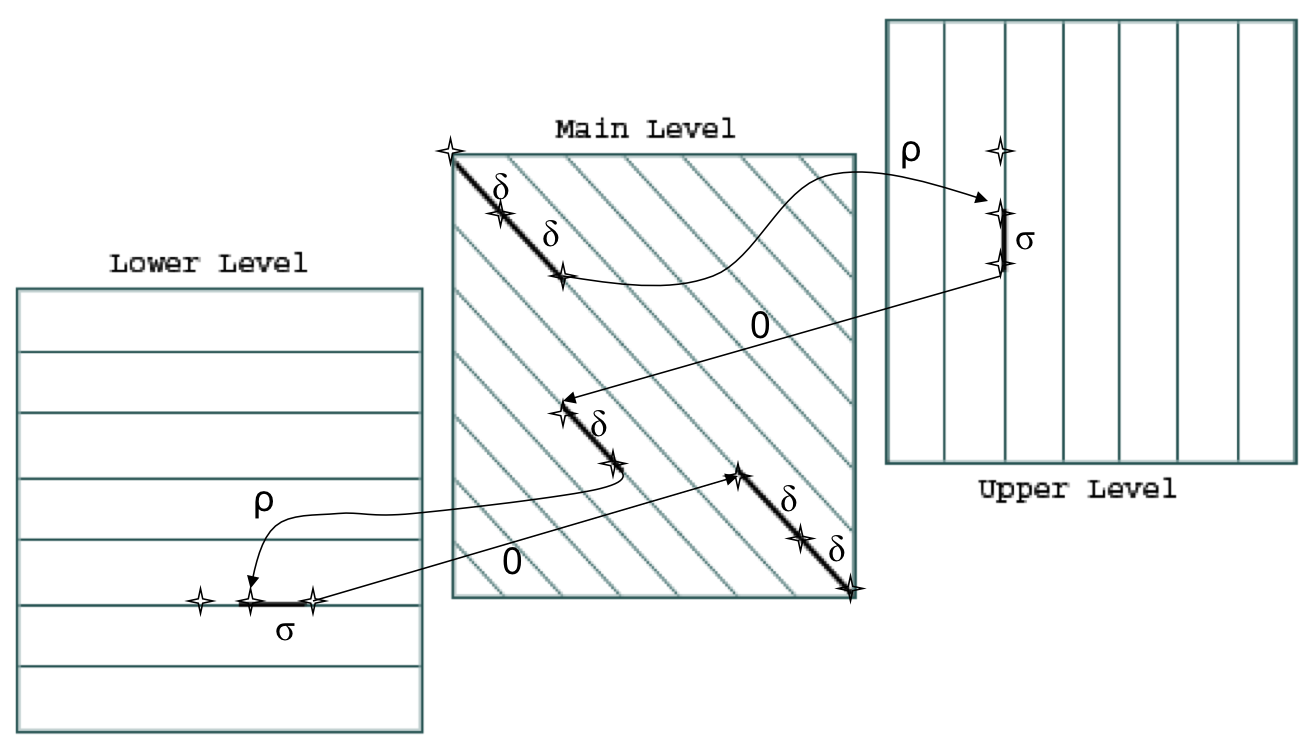
Levels:
- The main level is for diagonal edges
- The lower level is for horizontal edges
- The upper level is for vertical edges
A jumping penalty is assigned to moving from the main level to either the upper level or the lower level (-ρ - σ)
There is a gap extension penalty for each continuation on a level other than the main level (-σ)
12
Multiple Alignment versus Pairwise Alignment¶
 |  |
 |  |
- Up until now we have only tried to align two sequences.
- What about more than two? And what for?
- A faint similarity between two sequences becomes significant if present in many
- Multiple alignments can reveal subtle similarities that pairwise alignments do not reveal
13
Generalizing Pairwise Alignment¶
- Alignment of 2 sequences is represented as a 2-row matrix
In a similar way, we represent alignment of 3 sequences as a 3-row matrix
A T _ G C G _ A _ C G T _ A A T C A C _ A
- Score: more conserved columns, better alignment
14
Three-D Alignment Paths¶
- An alignment of 3 sequences: ATGC, AATC, ATGC
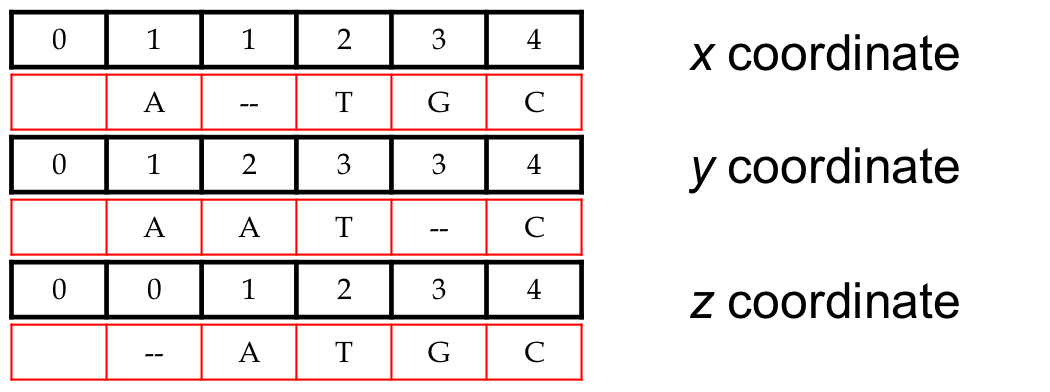
- Resulting path in (x,y,z) space:
(0,0,0) → (1,1,0) → (1,2,1) → (2,3,2) → (3,3,3) → (4,4,4) - Is there a better one?
15
Aligning Three Sequences¶
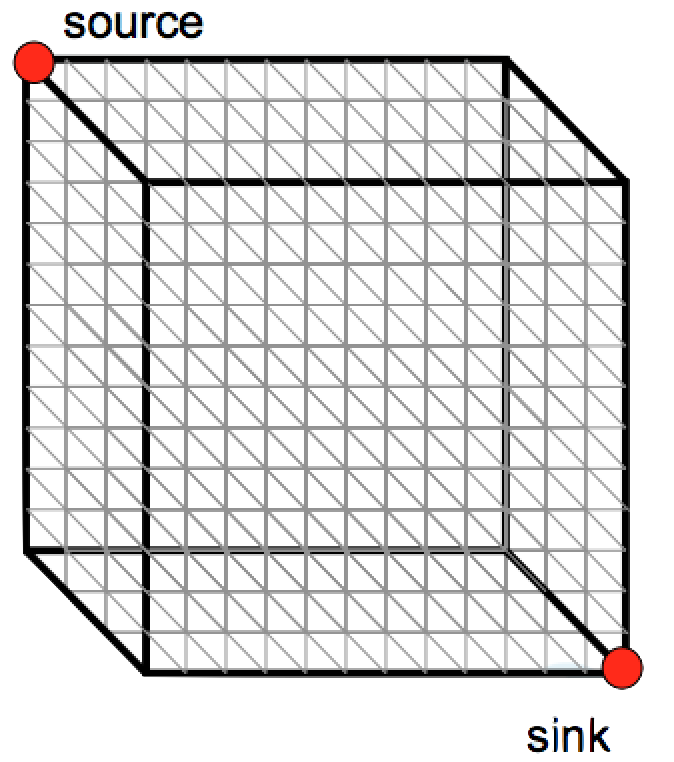
- Same strategy as aligning two sequences
- Use a 3-D “Manhattan Cube”, with each axis representing a sequence to align
- For global alignments, go from source to sink
16
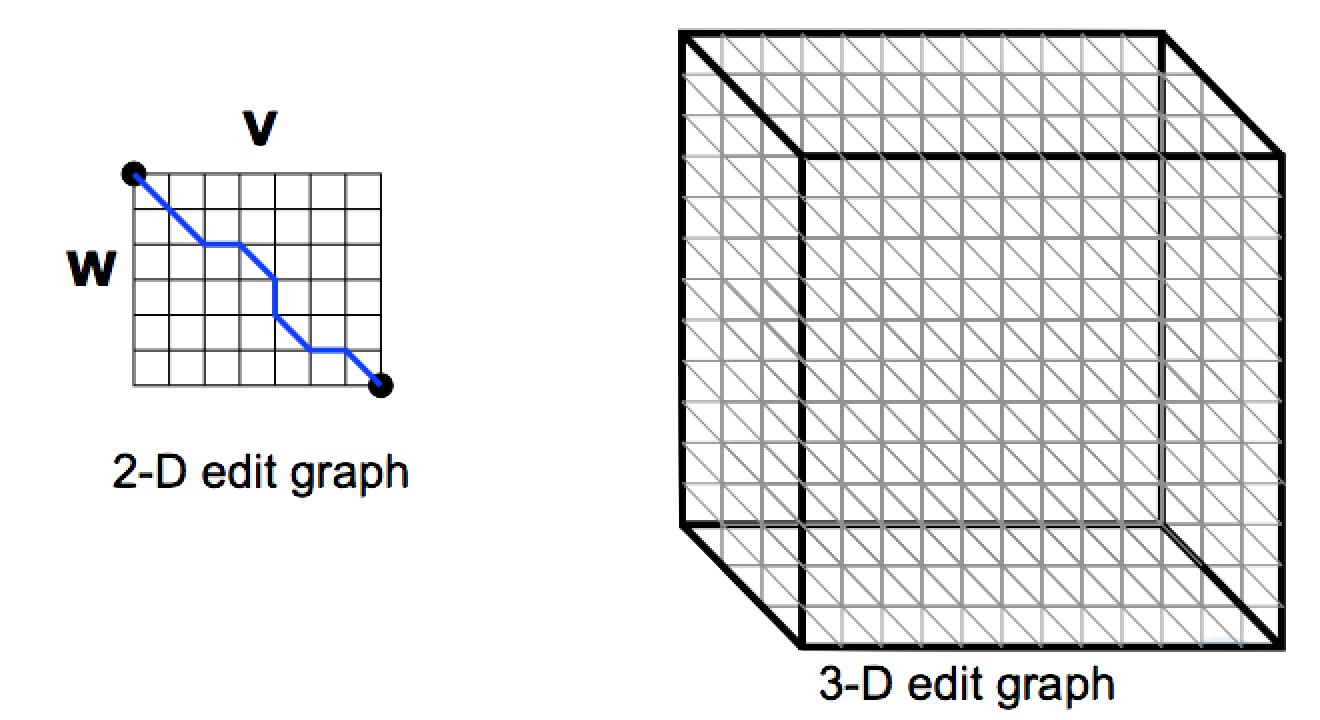
A 2-D cell versus a 3-D Alignment Cell¶
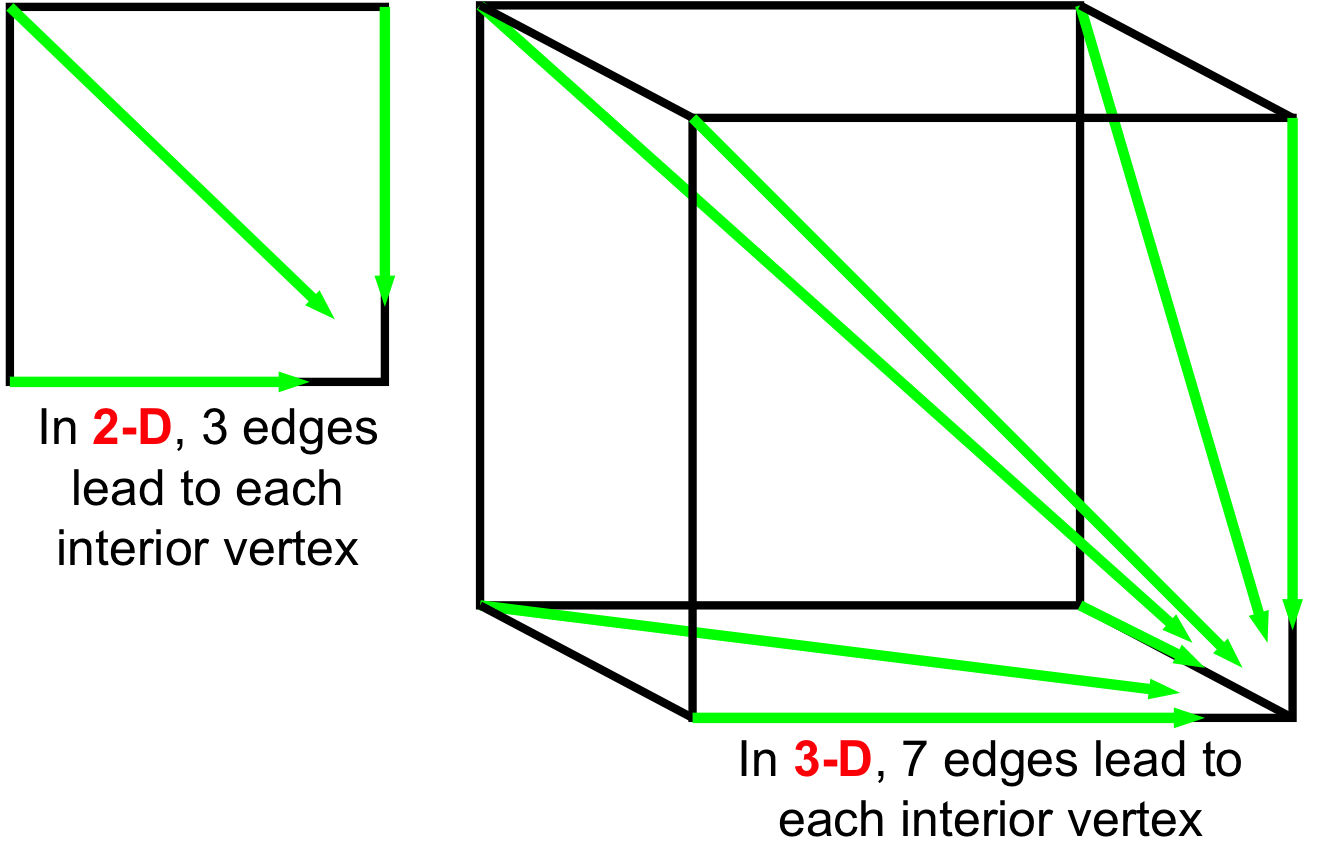
- 2-D [(i-1,j-1), (i-1,j), (i,j-1)] → (i,j)
- 3-D [(i-1,j-1,k-1), (i-1,j,k), (i,j-1,k), (i,j,k-1), (i,j-1,k-1), (i-1,j,k-1), (i-1,j-1,k),] → (i,j,k)
18
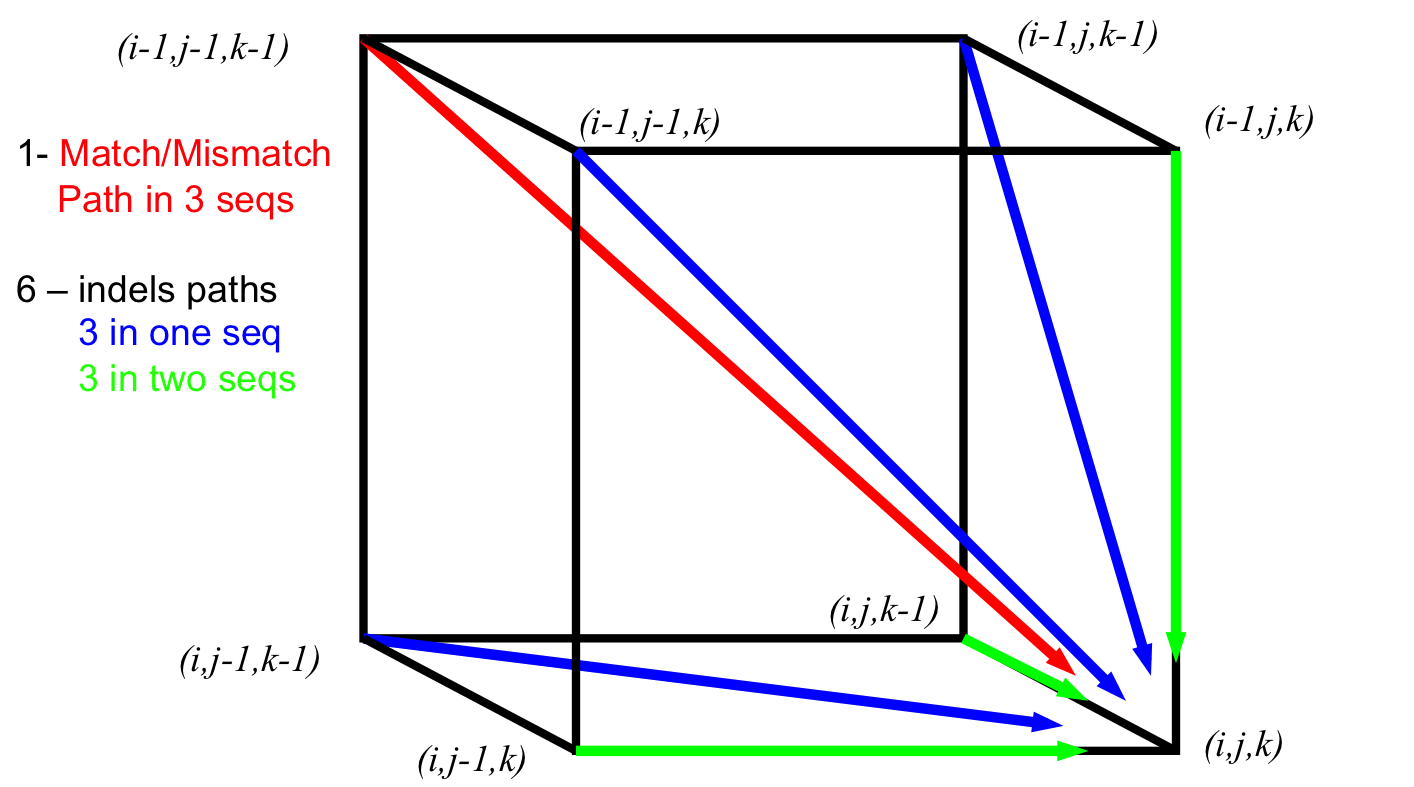
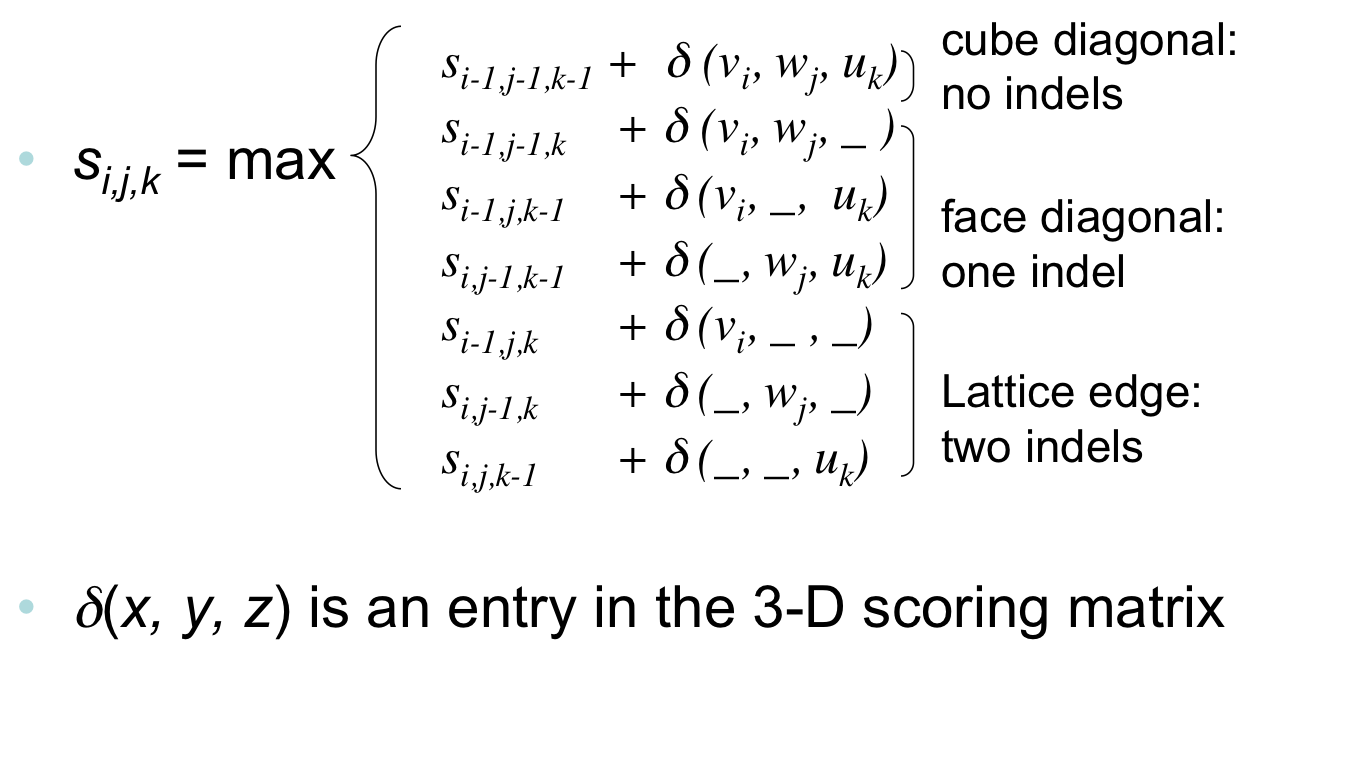
Multiple Alignment: Running Time¶
For 3 sequences of length n, the run time is $7n^3$; $O(n^3)$
For k sequences, build a k-dimensional Manhattan, with run time $(2^k-1)(n^k)$; $O(2^kn^k)$
Conclusion: dynamic programming approach for alignment between two sequences is easily extended to k sequences but it is impractical due to exponential running time
21
Multiple Alignment Induces Pairwise Alignments¶
Every multiple alignment induces pairwise alignments
x: AC-GCGG-C
y: AC-GC-GAG
z: GCCGC-GAG
Induces:
x: ACGCGG-C; x: AC-GCGG-C; y: AC-GCGAG
y: ACGC-GAC; z: GCCGC-GAG; z: GCCGCGAG
22
Inverse Problem¶
Do Pairwise Alignments imply a Multiple Alignment?
Given 3 arbitrary pairwise alignments:
x: ACGCTGG-C; x: AC-GCTGG-C; y: AC-GC-GAG y: ACGC--GAC; z: GCCGCA-GAG; z: GCCGCAGAG
Can we construct a multiple alignment that induces them?
NOT ALWAYSWhy? Because pairwise alignments may be arbitrarily inconsistent
23
Combining Optimal Pairwise Alignments¶
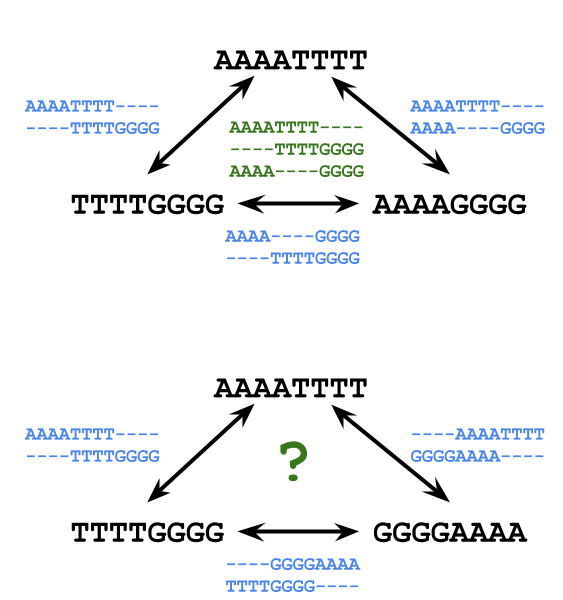
- In some cases we can combine pairwie alignments into a single multiple alignment
But, in others we cannot because one alignment makes a choice that is inconsistent with the overall best choice
AAAATTTT-------- ----AAAATTTT---- ----TTTTGGGG---- -OR- --------TTTTGGGG --------GGGGAAAA GGGGAAAA--------Is there another way?
24
Multiple Alignment from Pairwise Alignments¶
- From an optimal multiple alignment, we can infer pairwise alignments between all pairs of sequences, but they are not necessarily optimal
- It is difficult to infer a “good” multiple alignment from optimal pairwise alignments between all sequences
- Are we stuck, or is there some other trick?
25
Multiple Alignment using a Profile Scores¶
We used profile scores earlier when we discussed Motif finding
- A G G C T A T C A C C T G T A G – C T A C C A - - - G C A G – C T A C C A - - - G C A G – C T A T C A C – G G C A G – C T A T C G C – G G A 0 5 0 0 0 0 5 0 0 4 0 0 0 0 C 3 0 0 0 5 0 0 2 5 0 3 1 0 0 G 0 0 5 1 0 0 0 0 0 1 0 0 2 5 T 1 0 0 0 0 5 0 3 0 0 0 0 1 0 - 1 0 0 4 0 0 0 0 0 0 2 4 2 0Thus far we have aligned sequences against other sequences
Can we align a sequence against a profile?
Can we align a profile against a profile?
26
Aligning Alignments¶
A more general version of the multi-alignment problem:
Given two alignments, can we align them?
x: GGGCACTGCAT y: GGTTACGTC-- Alignment 1 z: GGGAACTGCAG w: GGACGTACC-- Alignment 2 v: GGACCT-----Idea: don’t use the sequences, but align their profiles
x: GGGCAC=TGCAT y: GGTTAC=GTC-- z: GGGAAC=TGCAG Combined Alignment || || | | w: GG==ACGTACC-- v: GG==ACCT-----
27
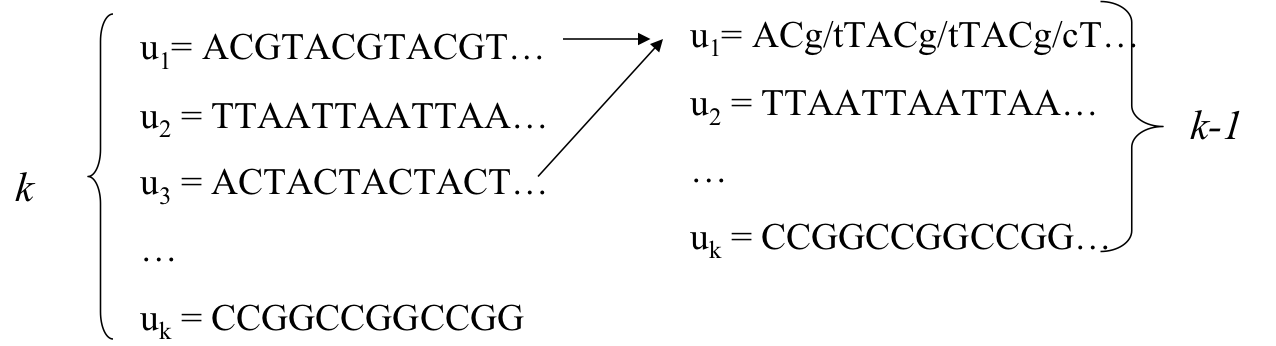
Profile-Based Multiple Alignment: A Greedy Approach¶
- Choose the most similar pair of strings and combine them into a profile, thereby reducing alignment of k sequences to an alignment of of k-1 sequences/profiles. Repeat
- This is a heuristic greedy method
28
Example¶
- Consider these 4 sequences
s1: GATTCA
s2: GTCTGA
s3: GATATT
s4: GTCAGC
- with the scoring matrix: {Match = 1, Mismatch = -1, Indel = -1}
29
Example (continued)¶
- There are ${4 \choose 2} = 6$ possible pairwise alignments
s2: GTCTGA s1: GATTCA--
s4: GTCAGC (score = 2) s4: G-T-CAGC (score = 0)
s1: GAT-TCA s2: G-TCTGA
s2: G-TCTGA (score = 1) s3: GATAT-T (score = -1)
s1: GAT-TCA s3: GAT-ATT
s3: GATAT-T (score = 1) s4: G-TCAGC (score = -1)
- The best pairwise score, 2, is between s2 and s4
29
Example (continued)¶
- Combine s2 and s4:
s2: G T C T G A
| | | | → s2,4: G T C t/a G a/c
s4: G T C A G C
- Giving a set of three sequences:
s1 : G A T T C A
s3 : G A T A T T
s2,4: G T C t/a G a/c
- Repeat for ${3 \choose 2} = 3$ possible pairwise alignments
s1 : GAT-TCA
s3 : GATAT-T (score = 1 + 1 + 1 - 1 + 1 - 1 - 1 = 1)
s1 : GAT-TCA
s2,4: G-TCtGa (score = 2 - 2 + 2 - 2 + 1 - 1 + 1 = 1)
s3 : GATAT-T
s2,4: G-TCtGa (score = 2 - 2 + 2 - 2 + 1 - 1 - 1 = -1)
29
Progressive Alignment¶
Progressive alignment is a variation of a greedy profile alignment algorithm with a somewhat more intelligent strategy for choosing the order of alignments.
Progressive alignment works well for close sequences, but deteriorates for distant sequences
- Once a gap appears in a consensus string it is permanent
- Uses profiles to compare sequences
CLUSTAL OMEGA
30
Clustal Omega¶
- A popular multiple alignment tool commonly used today
‘W’ stands for ‘weighted’ (different parts of alignment are weighted differently).
Three-step process
- Construct pairwise alignments
- Build Guide Tree
- Progressive Alignment guided by the tree
31
Clustal Omega's First Step¶
Pairwise alignment
- Align each sequence against all others giving a similarity matrix
- Similarity = exact matches / sequence length (percent identity)
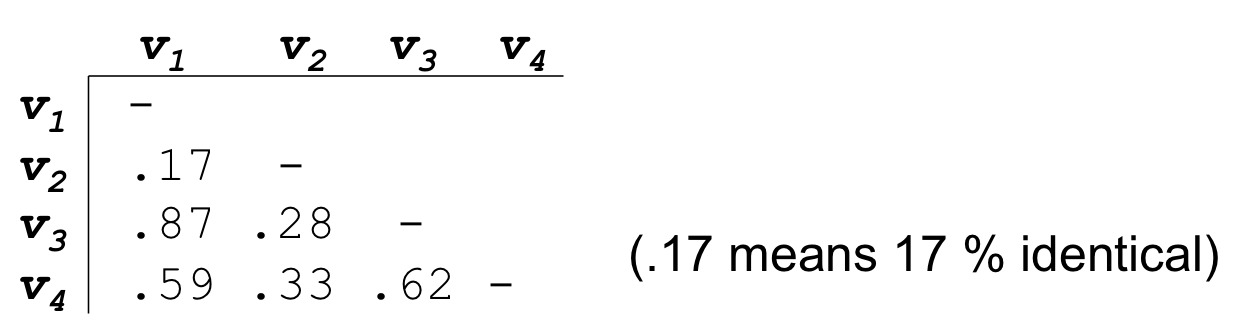
32
ClustalW's Second Step¶
- Create Guide Tree using the similarity matrix
- ClustalW uses the neighbor-joining method
(we will discuss this later in the course, in the section on clustering) - Guide tree roughly reflects evolutionary relations
- ClustalW uses the neighbor-joining method
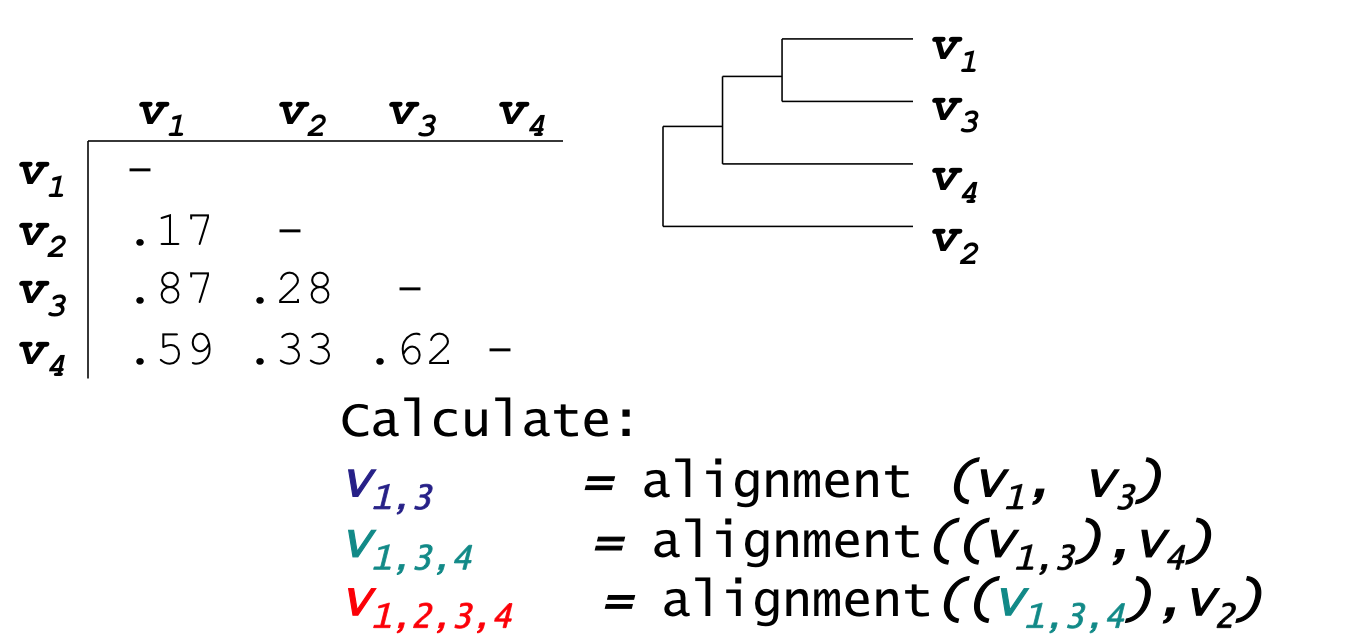
33
ClustalW's Third Step¶
- Start by aligning the two most similar sequences
- Following the guide tree, add in the next sequences, aligning to the existing alignment
- Insert gaps as necessary
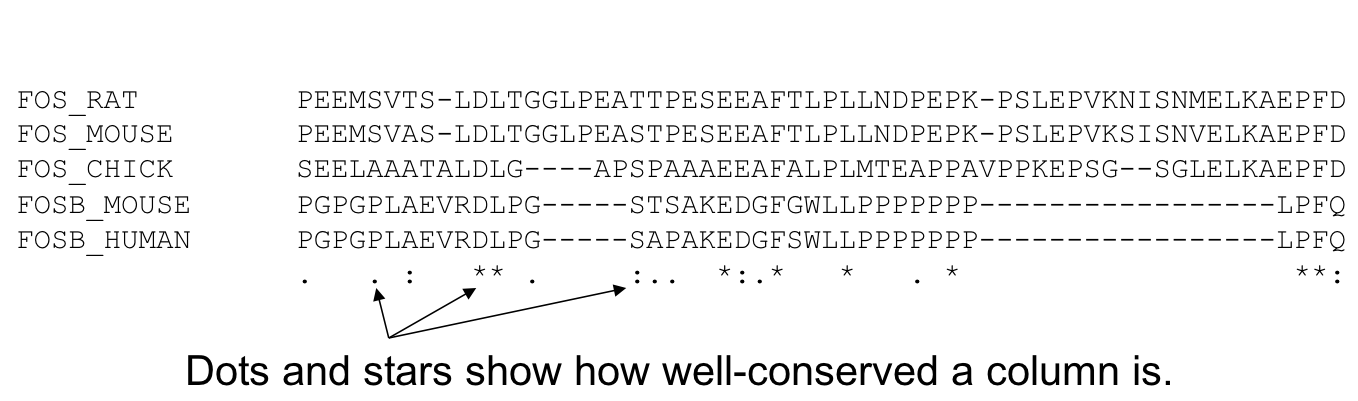
34
Next Time¶
- Midterm on Wednesday
- Covers material up to Lecture 11
- When we return from spring break we'll finish Sequence Alignment
35