Sequence Alignment¶

- Relating sequence alignment to our Manhattan Tour Problem
- Midterm exam (a week from Wednesday) covers up to this lecture
- No Class on Wednesday
1
A Biological Dynamic Programming Problem¶
- How to measure the similarity between a pair of nucleotide or amino acid sequences
- When Motif-Searching Problem we used Hamming distance as our measure
- Is Hamming distance the best measure?
- How can we distinguish matches that occur by chance from slightly modified patterns?
- What sorts of modifications should we allow?

2
Best Sequence Matches¶
- Depends on how you define Best
- Consider the two DNA sequences v and w :
u: ATATATAT
v: TATATATA
- The Hamming distance: dH(v, w) = 8 is large but the sequences are very similar
- What if we allowed insertions and deletions?
3
Allowing Insertions and Deletions¶
- By shifting one sequence over one position:
u: ATATATAT-
v: -TATATATA
- The edit distance: dH(v, w) = 2.
- Hamming distance neglects insertions and deletions
4
Edit Distance¶
Levenshtein (1966) introduced the notion of an “edit distance” between two strings as the minimum number of elementary operations (insertions, deletions, and substitutions) to transform one string into the other. (But, he gave no solution)
d(v,w) = Minimum number of elementary operations to transform v → v
Computing Hamming distance is a trivial task
Computing edit distance is non-trivial
5
Edit Distance: Example¶
TGCATAT → ATCCGAT in 5 steps
TGCATAT → (DELETE last T)
TGCATA → (DELETE last A)
TGCAT → (INSERT A at front)
ATGCAT → (SUBSTITUTE C for 3rd G)
ATCCAT → (INSERT G before last A)
ATCCGAT (Done)
What is the edit distance? 5?
6
Edit Distance: Example (2nd Try)¶
TGCATAT → ATCCGAT in 4 steps
TGCATAT → (INSERT A at front)
ATGCATAT → (DELETE 6th T)
ATGCAAT → (SUBSTITUTE G for 5th A)
ATGCGAT → (SUBSTITUTE C for 3rd G)
ATCCGAT (Done)
Is 4 the minimum edit distance? 3?
- A little jargon: Since the effect of insertion in one string can be accomplished via a deletion in the other string these two operations are quite similar. Often algorithms will consider them together as a single operation called INDEL
7
Longest Common Subsequence¶
- A special case of edit distance where no substitutions are allowed
- A subsequence need not be contiguous, but order must be preserved Ex. If v = ATTGCTA then AGCA and TTTA are subsequences of v, but TGTT and ACGA are not
- The length of the LCS, s, is related to the strings edit distance, d, by:
8
LCS as a Dynamic Program¶

- All possible possible alignments can be represented as a path from the string’s beginning (source) to its end (destination)
- Horizontal edges add gaps in v. Vertical edges add gaps in w. Diagonal edges are a match
- Notice that we’ve only included valid diagonal edges in our graph
9
Various Alignments¶
- Introduce coordinates for the grid
- All valid paths from the source to the destination represent some alignment

0 1 2 2 3 4 5 6 7 7
v A T _ G T T A T _
w A T C G T _ A _ C
0 1 2 3 4 5 5 6 6 7
Path: (0,0), (1,1), (2,2), (2,3), (3,4), (4,5), (5,5), (6,6), (7,6), (7,7)
10
Alternate Alignment¶
- Introduce coordinates for the grid
- All valid paths from the source to the destination represent some alignment

0 1 2 2 3 4 5 6 6 7
v A T _ G T T A _ T
w A T C G _ T A C _
0 1 2 3 4 4 5 6 7 7
Path: (0,0), (1,1), (2,2), (2,3), (3,4), (4,4), (5,5), (6,6), (6,7), (7,7)
11
Even Bad Alignments¶
- Introduce coordinates for the grid
- All valid paths from the source to the destination represent some alignment
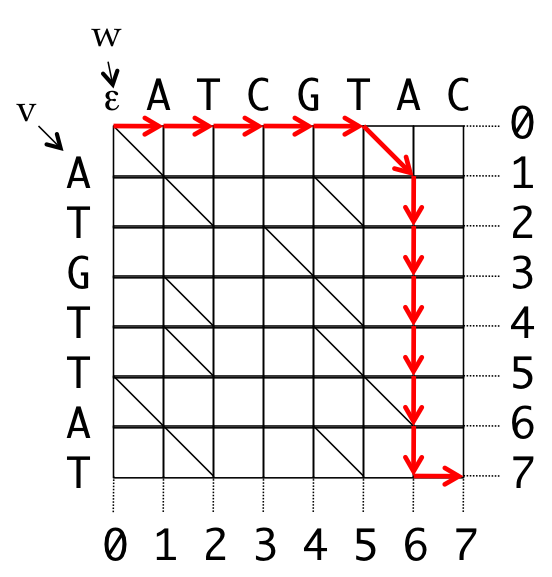
0 0 0 0 0 0 1 2 3 4 5 6 7 7
v _ _ _ _ _ A T G T T A T _
w A T C G T A _ _ _ _ _ _ T
0 1 2 3 4 5 6 6 6 6 6 6 6 7
Path: (0,0), (0,1), (0,2), (0,3), (0,4), (0,5), (1,6),
(2,6), (3,6), (4,6), (5,6), (6,6), (7,6), (7,7)
12
What makes a Good Alignment?¶
- Use as many diagonal segments (matches) as possible
- The end of a good alignment from (j...k) begins with a good alignment from (i..j)
- Same as Manhattan Tourist problem, where the sites are only on the diagonal streets!
- Set diagonal street weights = 1, and horizontal and vertical weights = 0
13
Alignment: Dynamic Program¶


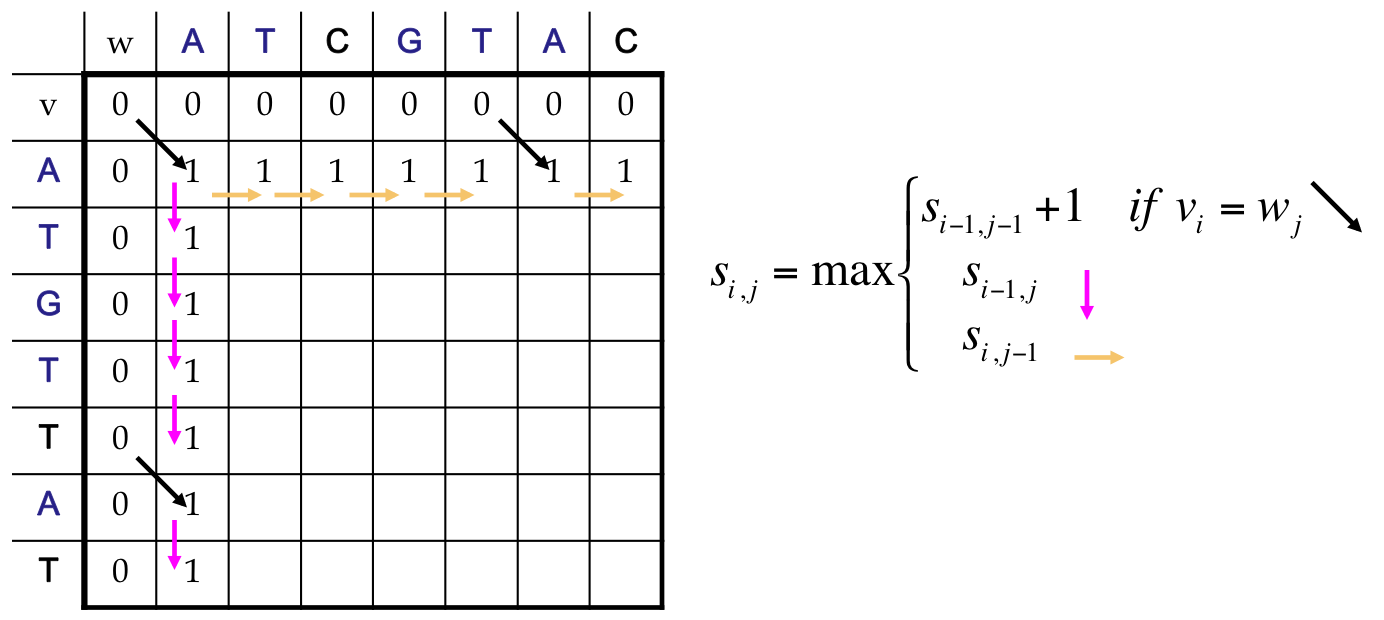
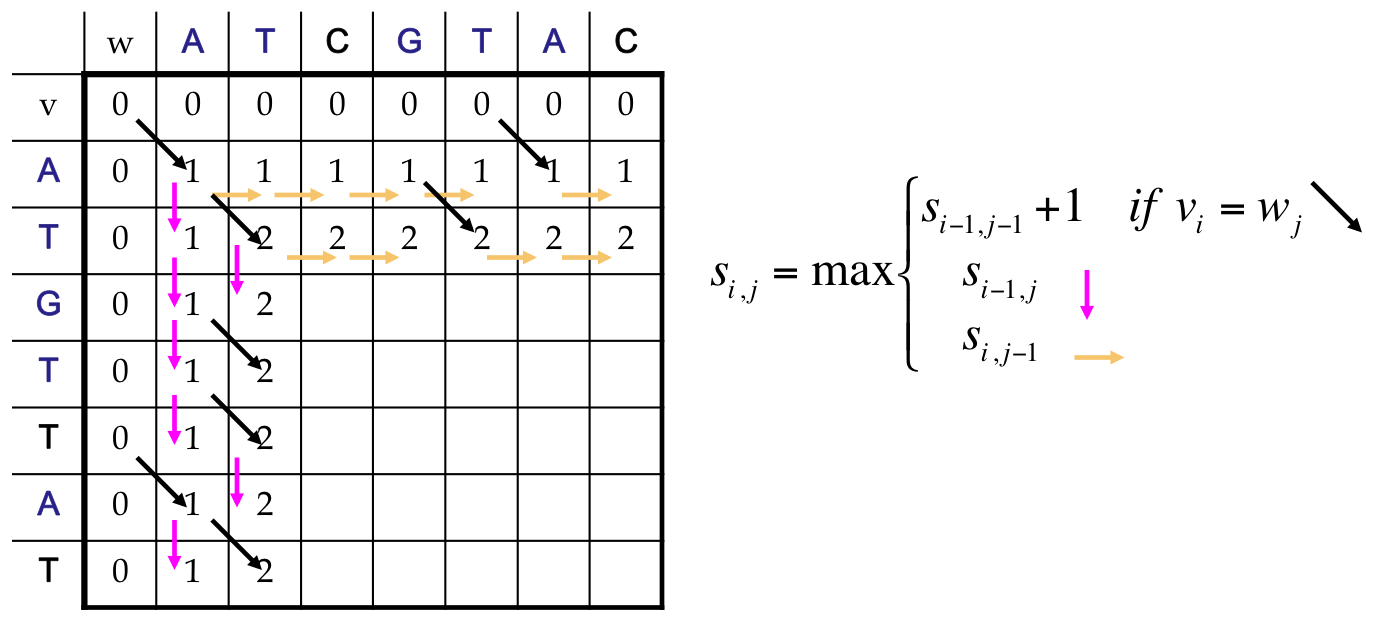
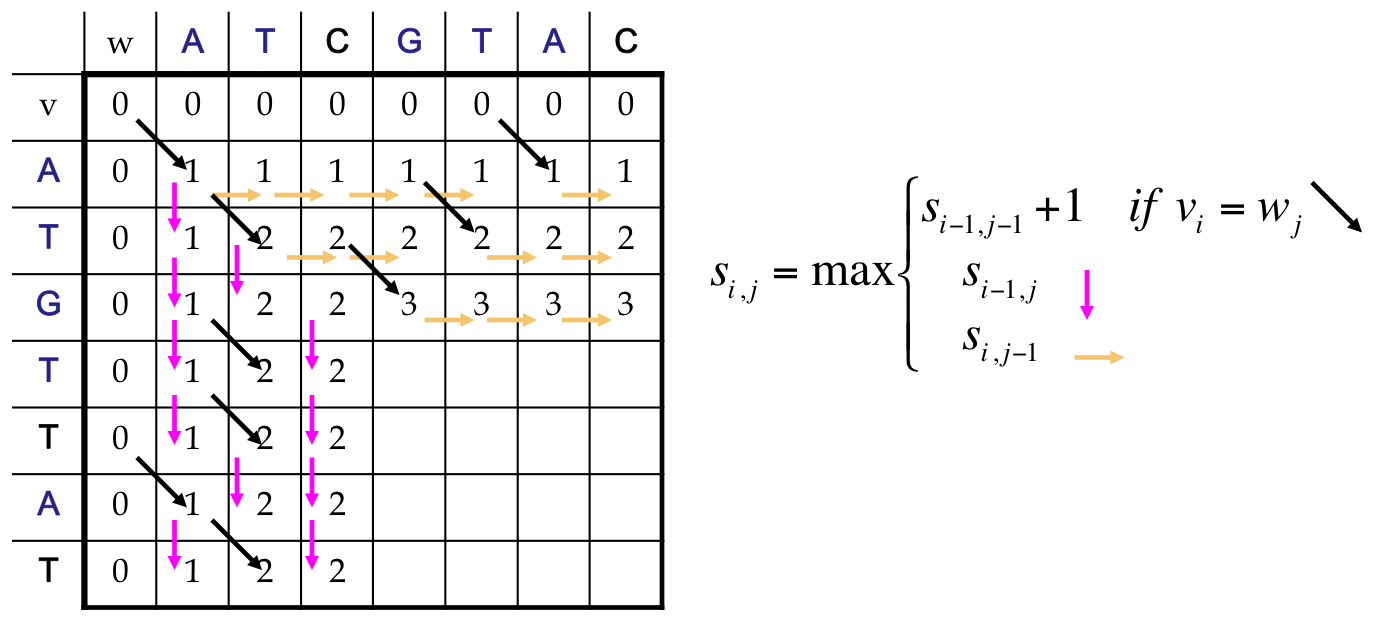
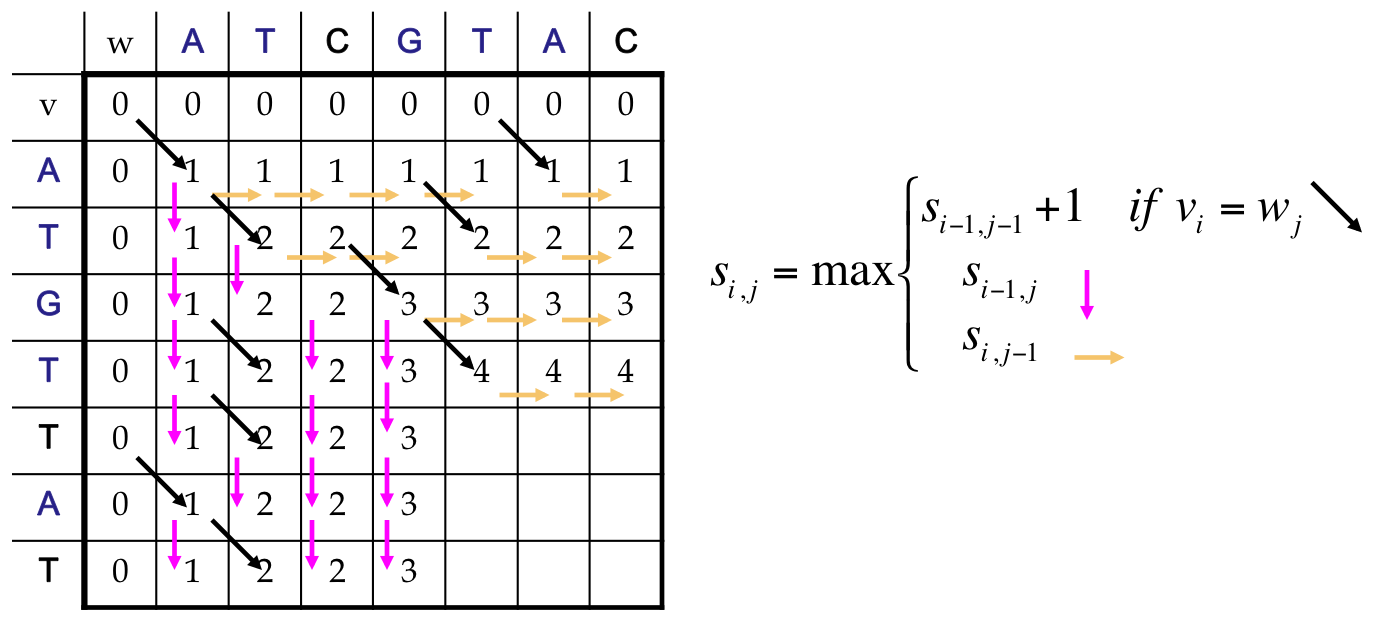
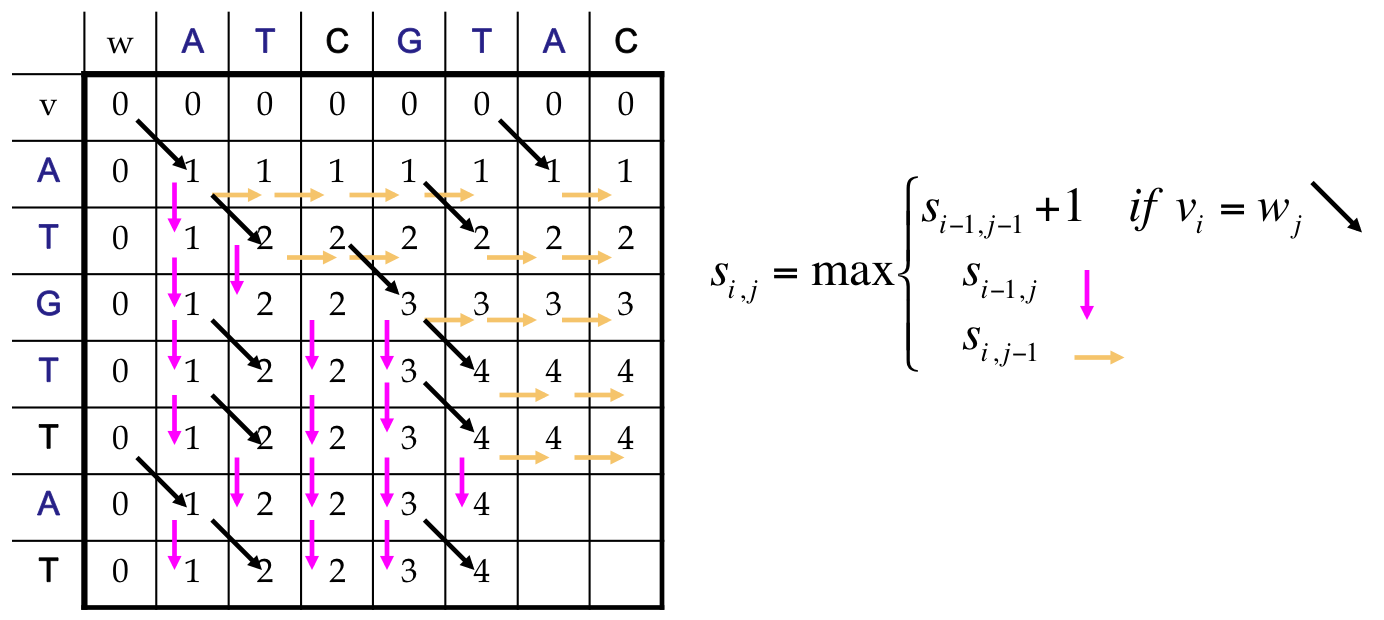
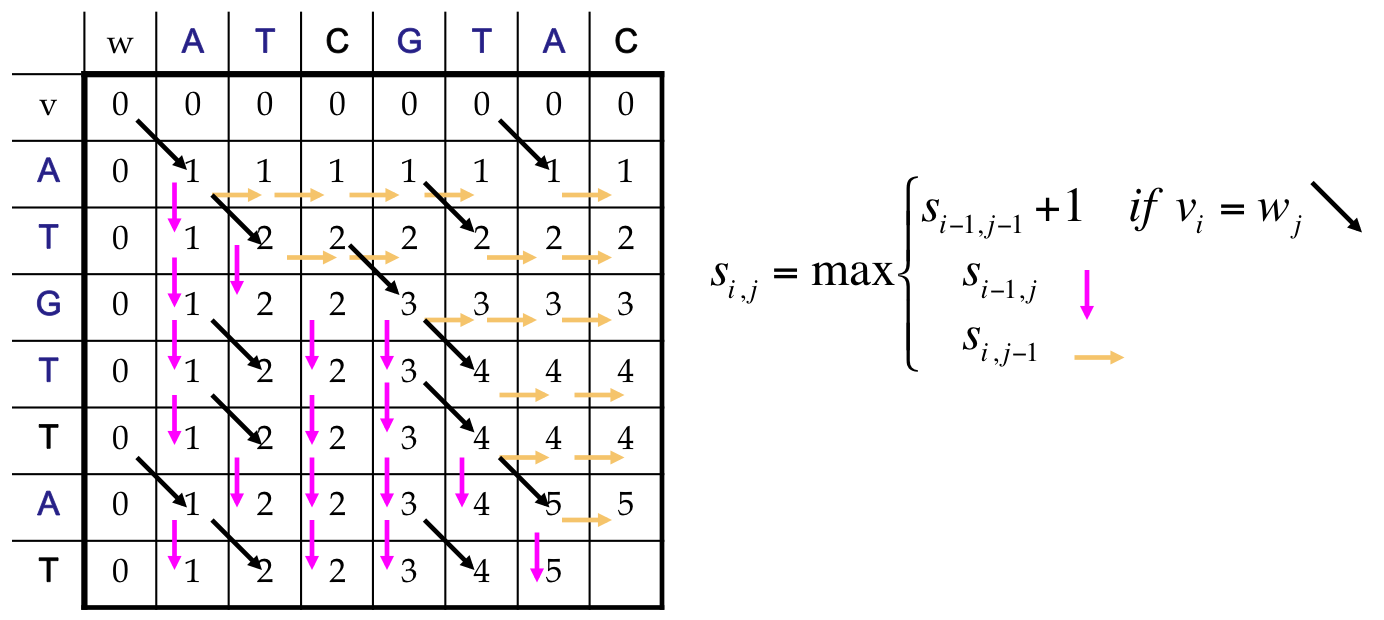
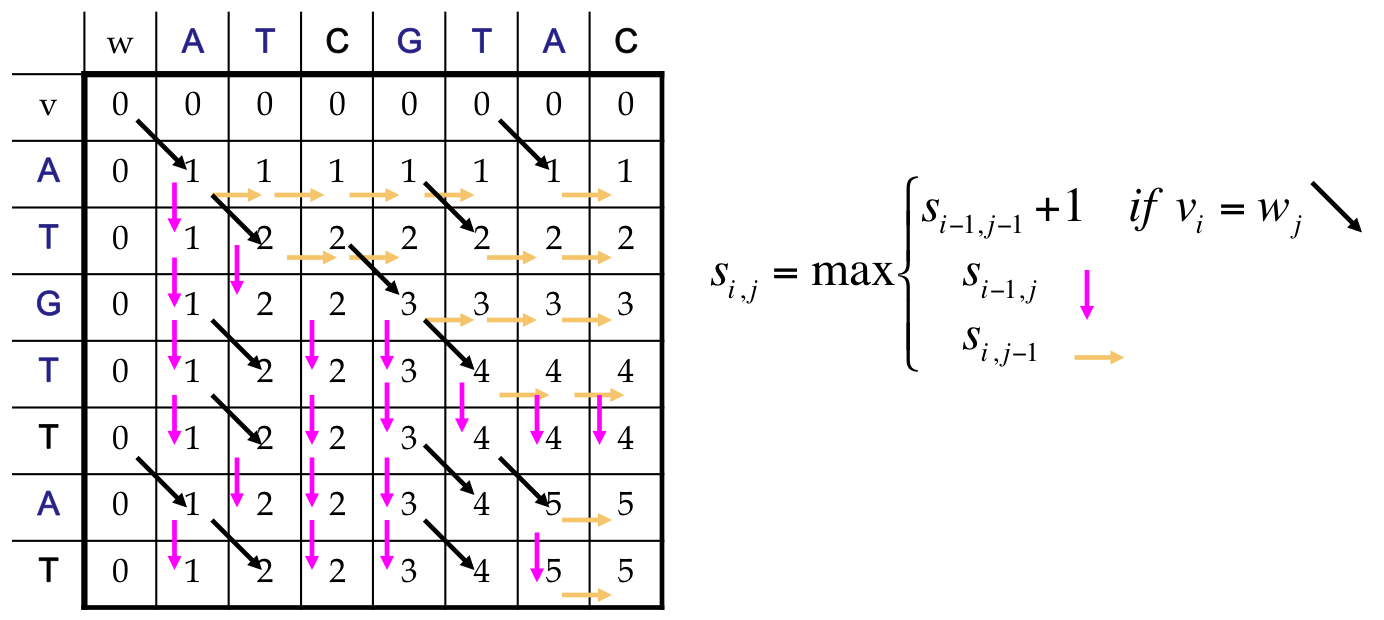
Finally¶
We reach the end, which corresponds to an LCS of length 5
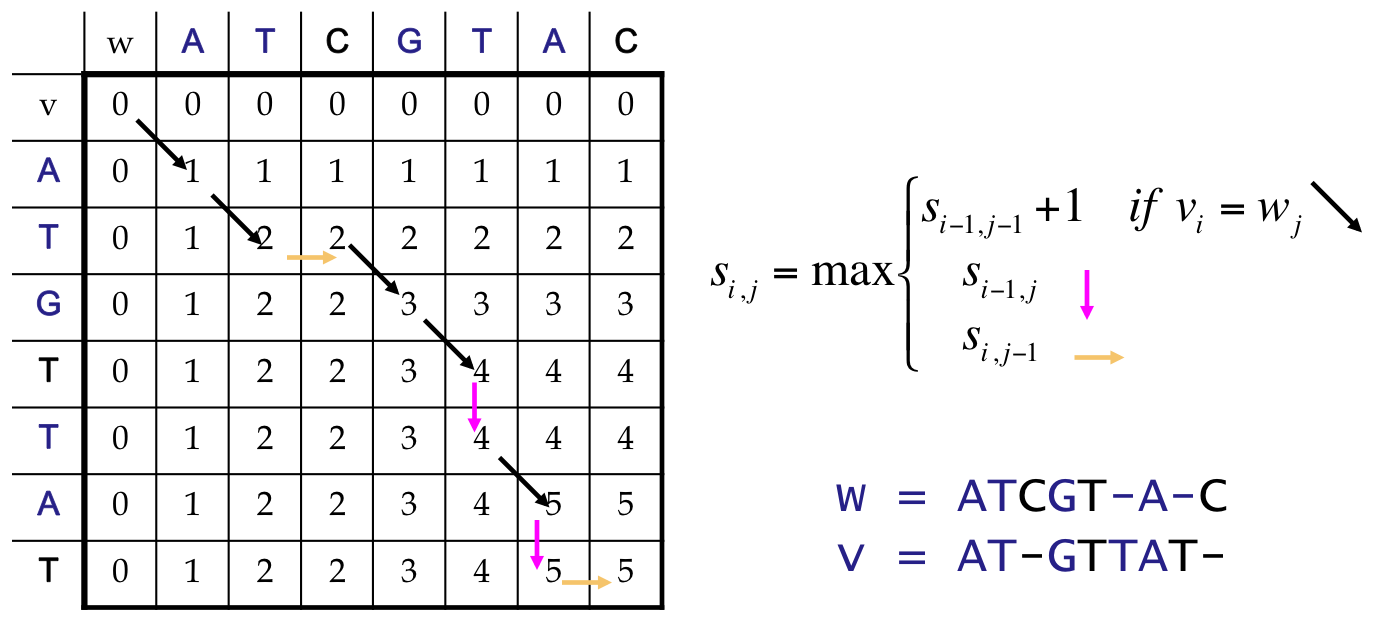
Our answer includes both an optimal score, and a path back to find the alignment
14
LCS Code¶
Let's see how well the code matches the approach we sketched out...
from numpy import *
def findLCS(v, w):
score = zeros((len(v)+1,len(w)+1), dtype="int32")
backt = zeros((len(v)+1,len(w)+1), dtype="int32")
for i in xrange(1,len(v)+1):
for j in xrange(1,len(w)+1):
# find best score at each vertex
if (v[i-1] == w[j-1]):
score[i,j], backt[i,j] = max((score[i-1,j-1] + 1, 3),
(score[i-1,j],1),
(score[i,j-1],2))
else:
score[i,j], backt[i,j] = max((score[i-1,j],1),
(score[i,j-1],2))
return (score, backt)
s, b = findLCS("ATGTTAT","ATCGTAC")
print "score =\n", s
print "\nbacktrack =\n", b
- This is the same score matrix that we found by hand
- The second matrix keeps track of the arrows that we used
15
Backtracking¶

In our example we used arrows {↓, →, \}, which were represented in our matrix as {1,2,3} respectively. This numbering is arbitrrary, except that it does break ties in our implementation (matches > w deletions > w insertions).
Next we need code that finds a path from the end of our strings to the beginning using our arrow matrix
16
Code to get our answer¶
We can write a simple recursive routine to return along the path of arrows that led to our best score.
def LCS(b,v,i,j):
if ((i == 0) and (j == 0)):
return ''
if (b[i,j] == 3):
result = LCS(b,v,i-1,j-1)
result = result + v[i-1]
return result
else:
if (b[i,j] == 2):
return LCS(b,v,i,j-1)
else:
return LCS(b,v,i-1,j)
print LCS(b,"ATGTTAT",b.shape[0]-1,b.shape[1]-1)
Perhaps we would like to get more than just the LCS; for example, the correpsonding alignment.
def Alignment(b,v,w,i,j):
if ((i == 0) and (j == 0)):
return ['','']
if (b[i,j] == 3):
result = Alignment(b,v,w,i-1,j-1)
result[0] += v[i-1]
result[1] += w[j-1]
return result
else:
if (b[i,j] == 2):
result = Alignment(b,v,w,i,j-1)
result[0] += "-"
result[1] += w[j-1]
return result
else:
result = Alignment(b,v,w,i-1,j)
result[0] += v[i-1]
result[1] += "-"
return result
align = Alignment(b,"ATGTTAT","ATCGTAC",b.shape[0]-1,b.shape[1]-1)
print "v =", align[0]
print "w =", align[1]
17
Alignment with a Scoring Matrix¶
- Scoring matrices can be created based on biological evidence.
- Alignments can be thought of as two sequences that differ due to mutations.
- Some of these mutations are more common or have little effect on the protein’s function, therefore some mismatch penalties, δ(vi, wj), should be less harsh than others.
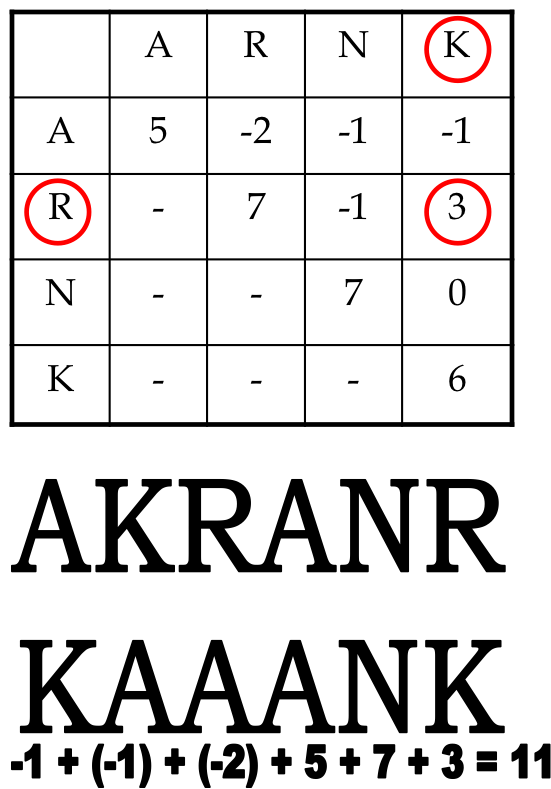
Example:
- Although R (arginine) and K (lysine) are different amino acids, they might still have a positive score.
- Why? They are both positively charged amino acids and hydrophillic implying such a substitution may not greatly change function of protein.
18
Functional Conservation¶
Amino acid changes that tend to preserve the electro-chemical properties of the original residue
- Polar to polar (aspartate → glutamate)
- Nonpolar to nonpolar (alanine → valine)
- Similarly behaving residues (leucine → isoleucine)
Common Amino acid substitution matrices
- PAM
- BLOSUM
DNA substitution matrices
- DNA is less conserved than protein sequences
- Less effective to compare coding regions at nucleotide level
19
PAM¶
- Point Accepted Mutation (Dayhoff et al.)
- 1 PAM = PAM1 = 1% average change of all amino acid positions
- After 100 PAMs of evolution, not every residue will have changed
- some residues may have mutated several times
- some residues may have returned to their original state
- some residues may not changed at all
- After 100 PAMs of evolution, not every residue will have changed
- PAMx ~ (PAM1)x
- PAM250 is a widely used scoring matrix
Ala Arg Asn Asp Cys Gln Glu Gly His Ile Leu Lys ...
A R N D C Q E G H I L K ...
Ala A 13 6 9 9 5 8 9 12 6 8 6 7 ...
Arg R 3 17 4 3 2 5 3 2 6 3 2 9
Asn N 4 4 6 7 2 5 6 4 6 3 2 5
Asp D 5 4 8 11 1 7 10 5 6 3 2 5
Cys C 2 1 1 1 52 1 1 2 2 2 1 1
Gln Q 3 5 5 6 1 10 7 3 7 2 3 5
...
Trp W 0 2 0 0 0 0 0 0 1 0 1 0
Tyr Y 1 1 2 1 3 1 1 1 3 2 2 1
Val V 7 4 4 4 4 4 4 4 5 4 15 10
20
BLOSUM¶
- Block Substitution Matrix
- Scores derived from observations of the frequencies of substitutions in blocks of local alignments in related proteins
- Matrix name indicates evolutionary distance
- BLOSUM50 was created using actual sequences sharing no more than 50% identity

21
Local vs. Global Alignment¶
The Global Alignment Problem tries to find the highest scoring path between vertices (0,0) and (n,m) in the edit graph.
The Local Alignment Problem tries to find the highest scoring subpath between all vertex pairs $(i_1,j_1)$ and $(i_2, j_2)$ in the edit graph where $i_2 > i_1$ and $j_2 > j_1$.
In an edit graph with negatively-weighted scores, a Local Alignment may score higher than a Global Alignment
Example:
Global Alignment
--T—-CC-C-AGT—-TATGT-CAGGGGACACG—A-GCATGCAGA-GAC | || | || | | | ||| || | | | | |||| | AATTGCCGCC-GTCGT-T-TTCAG----CA-GTTATG—T-CAGAT--CLocal Alignment finds longer conserved segment
tccCAGTTATGTCAGgggacacgagcatgcagagac |||||||||||| aattgccgccgtcgttttcagCAGTTATGTCAGatc
22
Local Alignments: Why?¶
- Two genes in different species may be similar over short conserved regions and dissimilar over remaining regions.
- Example:
- Homeobox genes have a short region called the homeodomain that is highly conserved between species.
- A global alignment would not find the homeodomain because it would try to align the ENTIRE sequence
Local Alignment Problem:
- Goal: Find the best local alignment between two strings
- Input: Strings v, w and scoring matrix δ
- Output: Alignment of substrings of v and w whose alignment score is maximum among all possible alignment of all possible substrings
23
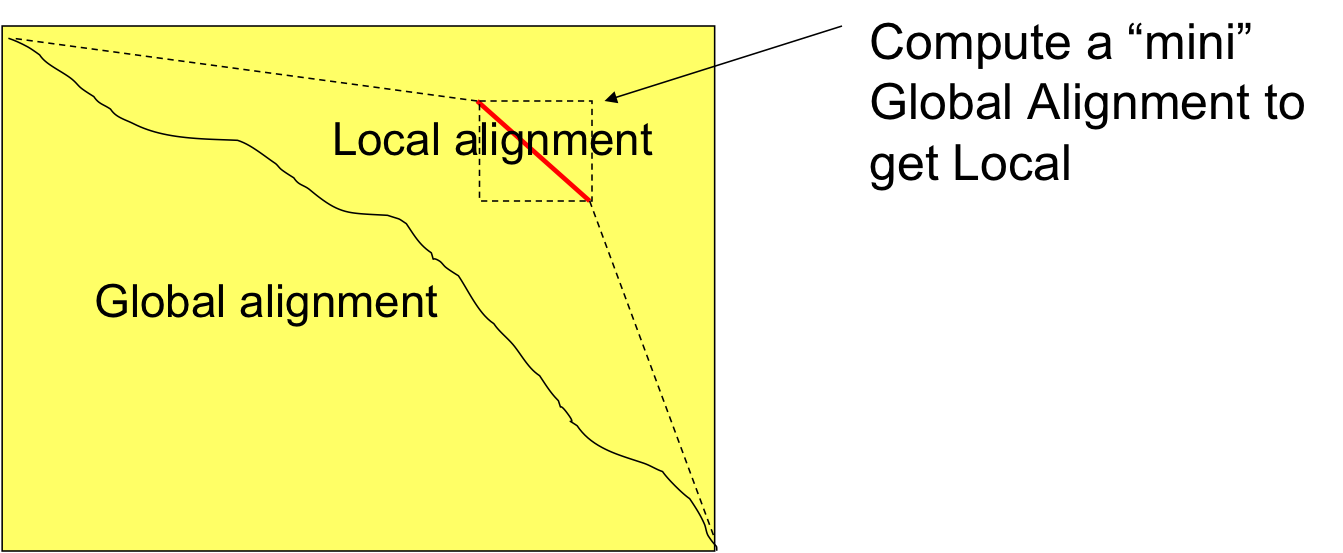
Brute Force Local Alignment¶
Find the best global alignment amoung all blocks $(i_1,j_1,i_2,j_2)$
Long run time $O(n^4)$:
- In the grid of size n x n there are $O(n^2)$ vertices $(i_1,j_1)$ that may serve as a source.
- For each such vertex computing alignments from $(i_1,j_1)$ to $(i_2,j_2)$ takes $O(n^2)$ time.
This can be remedied by giving free rides
25
Local Alignment: Free Rides¶
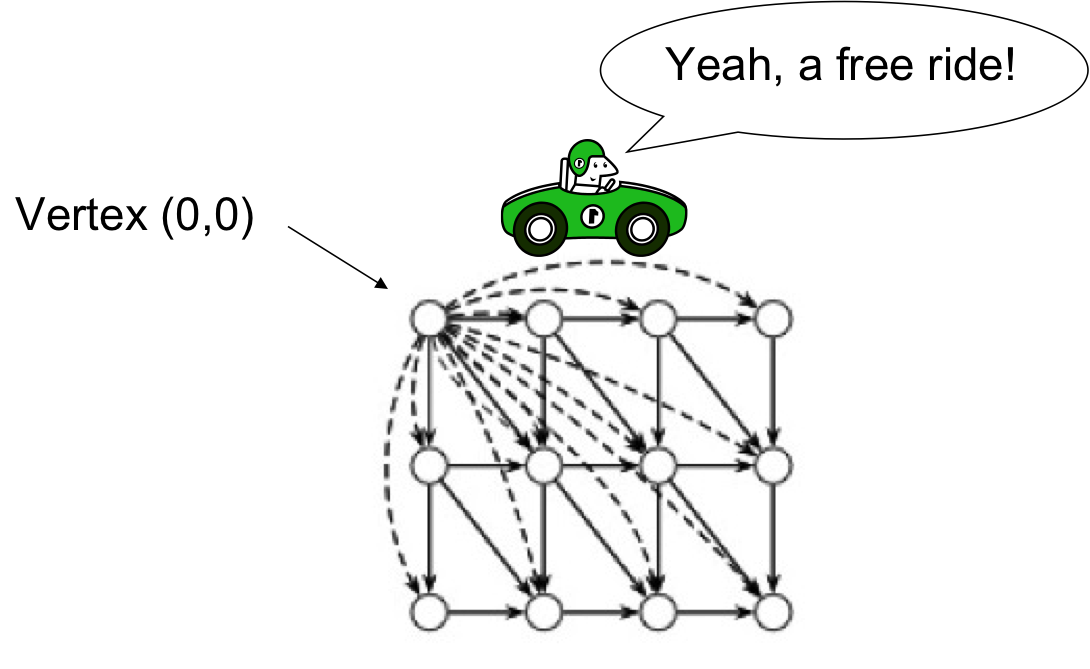
- The dashed edges represent a free ride from (0,0) to any other node
- The largest value of si,j over the whole score matrix is the end point of the best local alignment (instead of sn,m).
26
The Local Alignment Recurrence¶
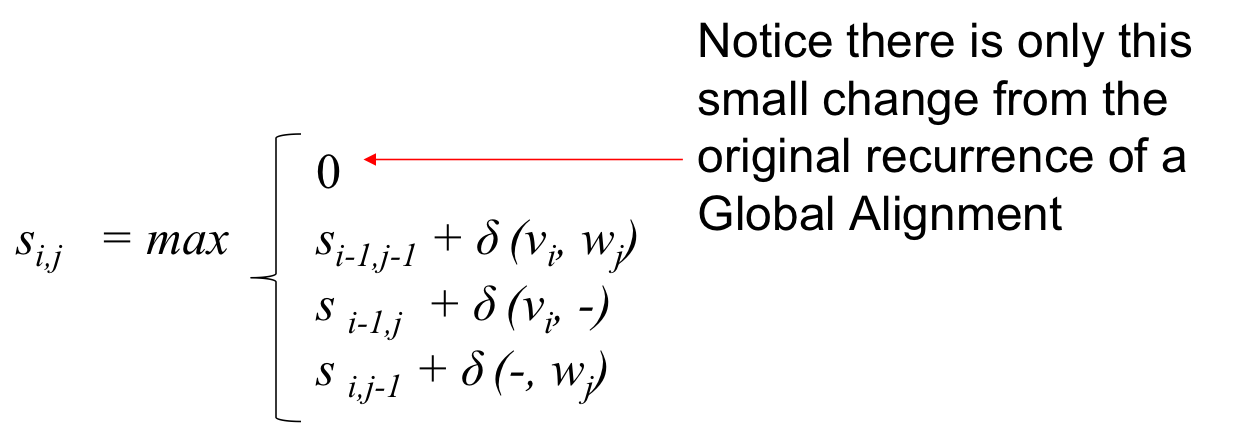
- The zero is our free ride that allows the node to restart with a score of 0 at any point
- What does this imply?
- After solving for the entire score matrix, we then search for si,j with the highest score, this is $(i_2,j_2)$
- We follow our back tracking matrix until we reach a score of 0, whose coordinate becomes $(i_1,j_1)$
27
Next Time¶
- Alignment with Gap Penalities
- Multiple Alignment problem
- Can we do better than $O(MN)$?
28