
What we know about Genomes¶
DNA sequences are a biological system's hard drive
- They contain an operating system with all the low-level support for growing, dividing, and reproducing
- They contain application programs for making cells that move our bodies, remember our mother's face, and store energy for use in lean times
- They are robust. They have programs for repairing and replicating themselves. They even have backups!
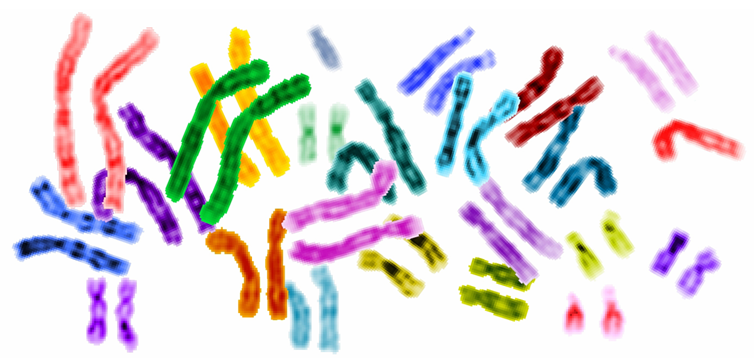
DNA sequences vary in size
- Human nuclear DNA is composed of roughly 6 billion base-pairs distrbuted over 46 pairs of chromosomes
- These 6 billion bases are comprised of 2 nearly identical copies
- One of these copies is called haplotype and its sequence is called a genome
- Among humans, any two haplotypes are are 99.9% identical
How can we read off the sequence of DNA?
2
DNA Sequencing History¶
- DNA sequencing was one of the most significant breakthroughs of the 20th century
- This was so inherently obvious it was awarded a Noble prize only 3 years after its development
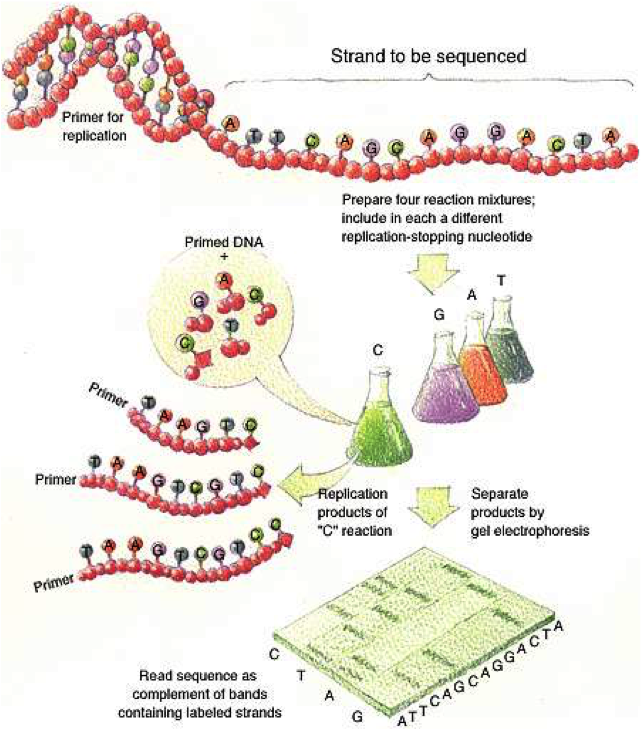
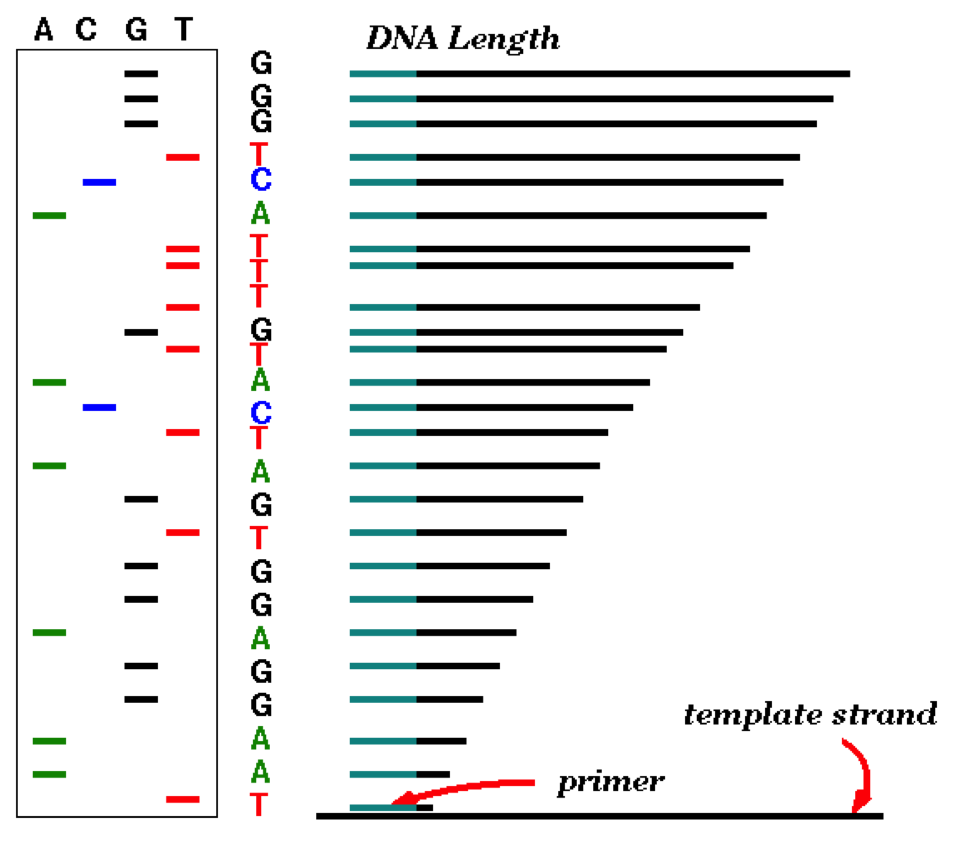
Sanger method (1977):
Labeled ddNTPs terminate DNA copying at random points.

Gilbert method (1977):
Chemical method to cleave DNA at specific points (G, G+A, T+C, C).

Both methods generate labeled fragments of varying lengths that are further electrophoresed
3
Sanger Method¶
|
  |
4
Assembling the Human Genome¶
In 1990, a moon-shot-like project was begun to sequence the entire Human Genome.
- It would require 30x coverage to provide enough sequences
- Recall there are sequence differences-- Approximately 1:1000 bases
- Redundacy was needed to find the majority base from 16 different individuals (32 genomes)
- Also needed the extra coverage to assure that there is enough overlap to assemble the 500 base-pair reads
A $3 billion dollar NIH funded public effort led by Francis Collins with a 15-year plan.
It would distribute the work across several labs in a community effort by assigning primers
to groups on a first-come basis. New sequencing results yielded new primers, so the project
required a central coordination.
|
In 1997 a private company, Celera, lead by Craig Venter, suggested they could beat the public effort by dispensing with primers.
They'd just randomly fragment DNA and sequence each fragment with no idea of how sequenced fragment would fit together. In other words, they were going to rely on computer science to assemble their reads algorithmically.

|
The result was that, despite tensions, the groups ended up sharing data and technologies. ANd the competition led to a completed draft 5 years ahead of schedule.
5
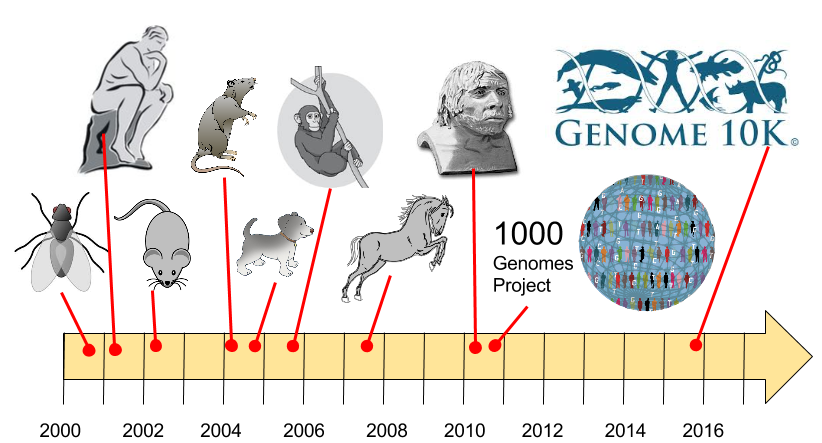
The Sequencing Race¶
Since the Human Genome project there have been an explosion of genomes sequenced. Initially, the focus was on model organisms, then favorites, then all of human diversity, and finally a catalog of life's diversity.
6
The secret behind this explosion of genomes¶
Next generation seqquencing machines have revolutionized the DNA sequencing process. They work in various ways including massiviely-parallel single-base extension methods, to captured Dnases whose motions suggest a the base being replicated, to microholes that only a single DNA molecule can pass through, and the bases are determined by detectable charge differences.

|
In a way, the *genome moonshot* was far more successful than the real moonshot. The rate at which genomes can be sequenced, and the cost per base has seen unprecented improvements. Faster than even Moore's Law.
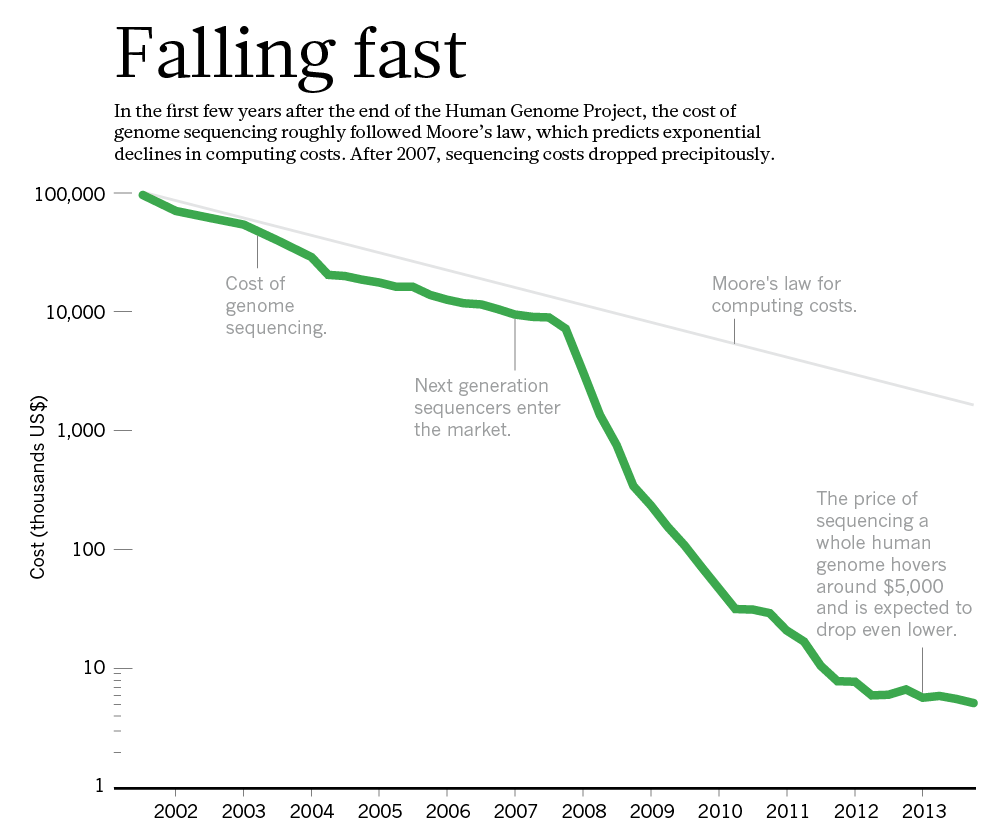
|
7
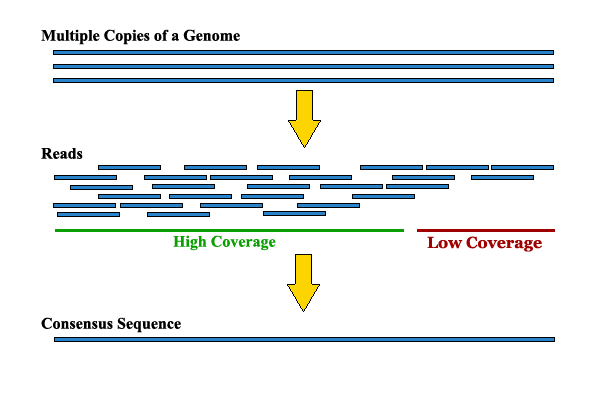
An Analogy¶

|

|

|

|
Some important differences
- A better analogy would have been to shred 100's of books
- Shuffle the pages before shredding
- Oh yeah, my book has approximately 850,000 characters.
- The entireity of Encyclopedia Britannica is approximately 250,000,000 characters. Your genome is approximately 12 times larger
9

Searching for overlaps¶

You'd look for fragments that fit together based on some overlapping context that they share.

And then, build upon those to assemble a more complete picture
11

The key idea was to link between a pair fragments¶
This leads us to a computational analogy called a graph
- A graph is composed of nodes, which can represent entities, in our case read fragments
- Nodes are connected by edges that represent some relationship between a pair of nodes
- The edges of a graph can be directed
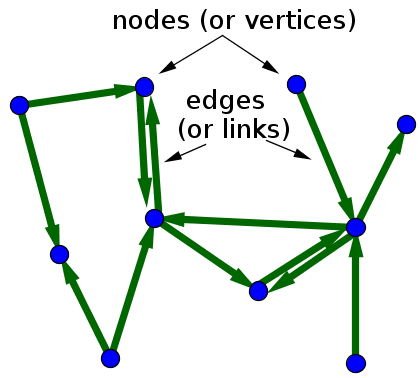
One can devise both representaions for, and algorithms that operate on, graphs.
- For example, you can find the shortest path between to nodes in a graph. Your GPS solves this problem, where addresses or locales are nodes, and roads are edges.
- You can find a minimal set of edges that maintains that keeps the graph connected
Let's rethink our DNA ssembly problem as a graph problem.
13
The graph of a sequence¶
For the moment let's imagine that reads are like k-mers from a sequence, as they do tend to be uniform in length.
GACGGCGGCGCACGGCGCAA - Our toy sequence
GACGG
ACGGC
CGGCG
GGCGG
GCGGC
CGGCG
GGCGC - The complete set of 16 5-mers
GCGCA
CGCAC
GCACG
CACGG
ACGGC
CGGCG
GGCGC
GCGCA
CGCAA
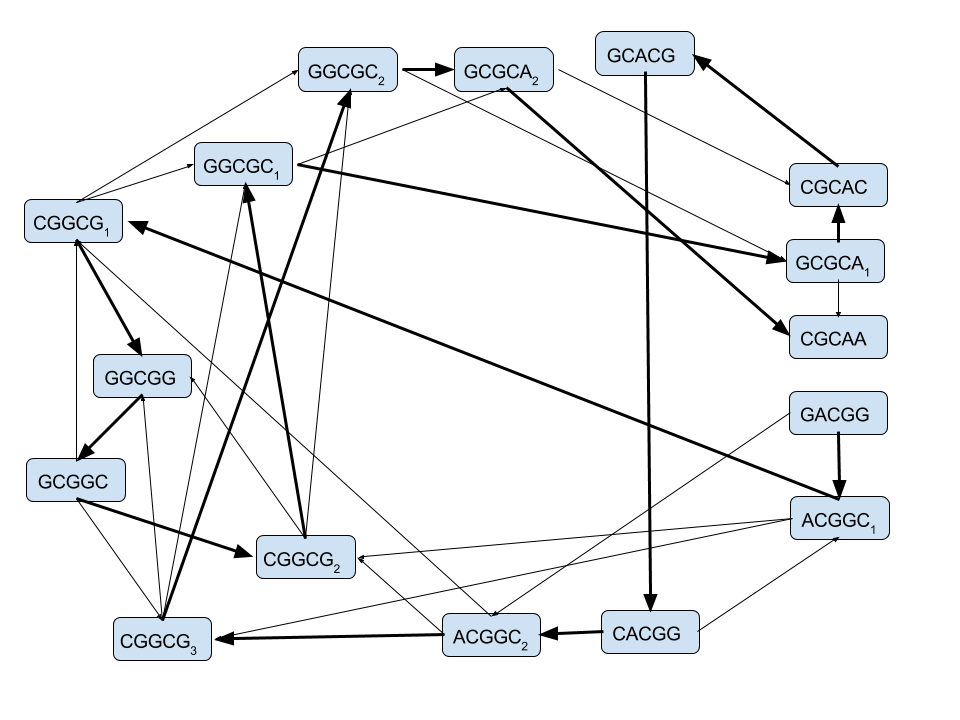
Now we can construct a graph where:
- Each 5-mer is a node
- There is a directed edge from a k-mer that shares its (k-1)-base suffix with the (k-1)-base prefix of another k-mer
14
A read-overlap graph¶
The read-overlap graph for the 5-mers from:
GACGGCGGCGCACGGCGCAA
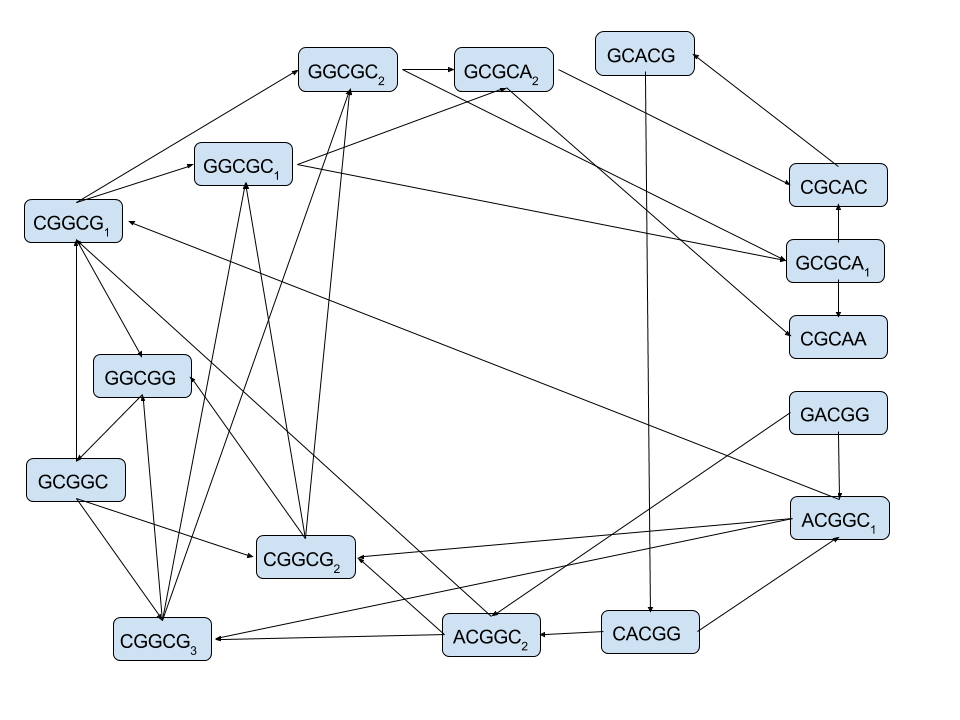
The problem is How to infer the original sequence from this graph?
15
The rules of our game¶
- Every node, k-mer, can be used exactly once
- The object is to find a path along edges that visits every node one time
- This game was invented in the mid 1800's by a mathematician called Sir William Hamilton
</ul>

A version of Hamilton's game: 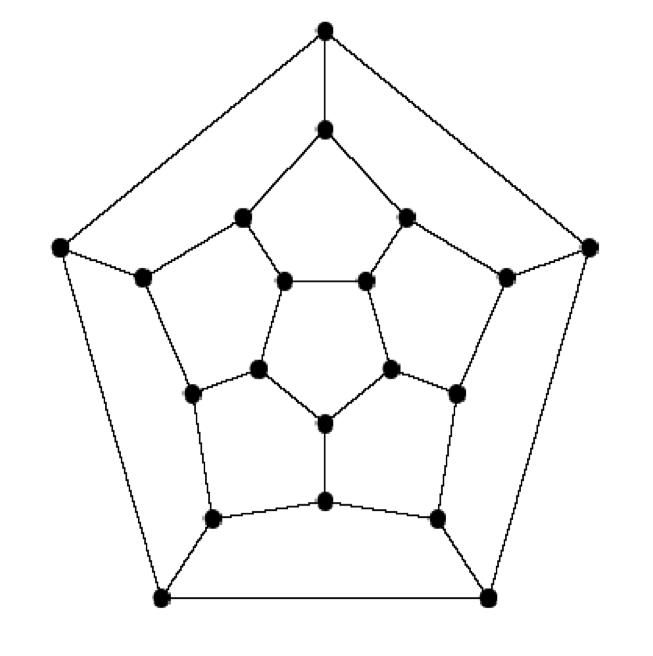
16
Finding a Hamiltonian Path in a graph¶
Our desired sequence:
GACGGCGGCGCACGGCGCAAis indeed a path in this graph
How can we write a program to solve Hamilton's puzzle?
Is the solution unique?
18
Another way that to represent our k-mers in a graph¶
- Rather than making each k-mer a node, let's try making them an edge
- That seems odd, but it is related to the overlap idea
- The 5-mer
GACGG
has a prefixGACG
and a suffixACGG
- Think of the k-mer as the edge connecting a prefix to a suffix
- This leads to a series of simple graphs
- The 5-mer
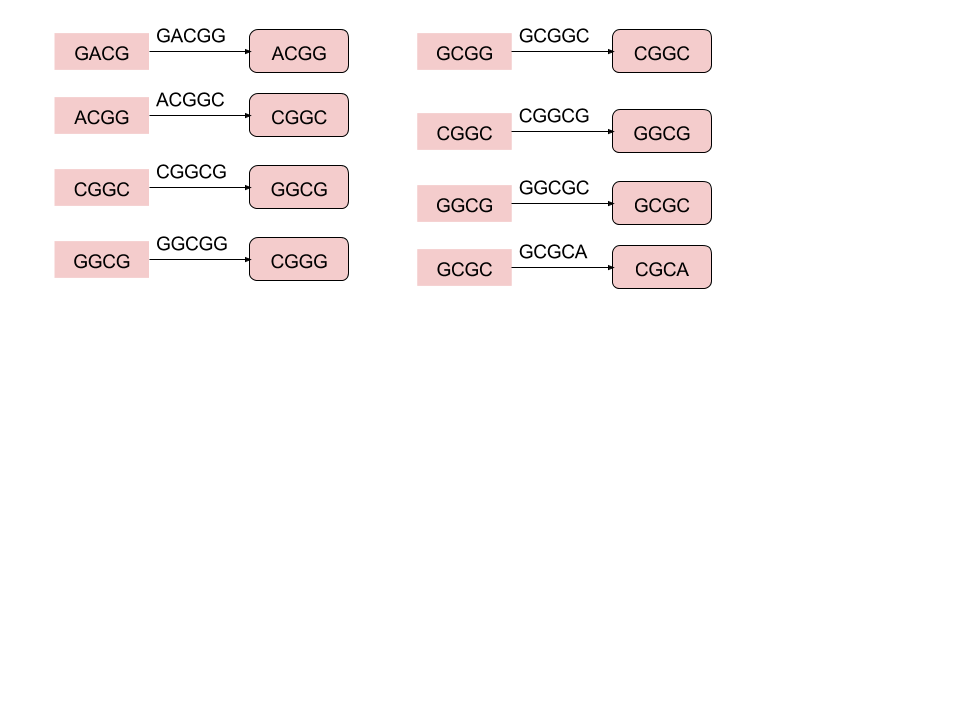
- Then combine all nodes with the same Label
19
A De Bruijn Graph¶
This rather odd graph is called the "De Bruijn" graph, was named after a famous mathematician.
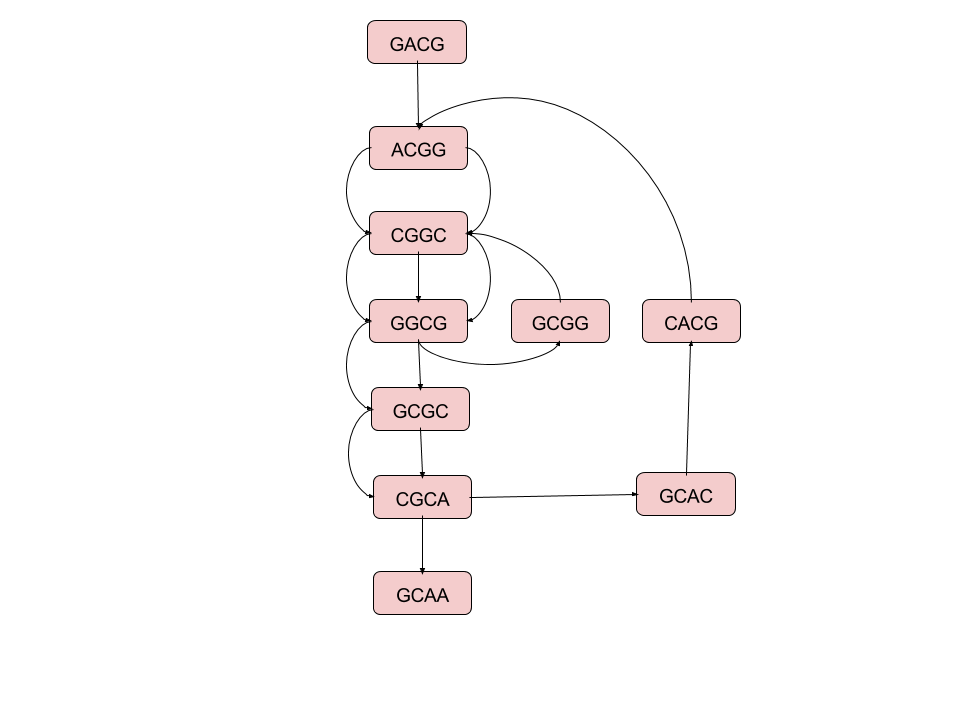
The problem is How to infer the original sequence from this graph?
20
The rules of our new game¶
- Every edge, k-mer, can be used exactly once
- The object is to find a path in the graph that uses each edge only one time
- This game was invented in the late 1700's by a mathematician called Leonhard Euler

Leonhard Euler |
A version of Euler's game:
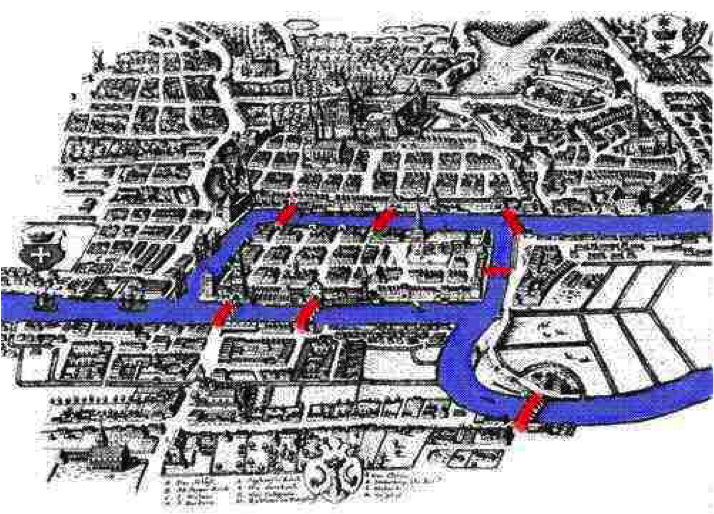 Bridges of Königsberg
Bridges of KönigsbergFind a city tour that crosses every bridge just once |
21
Next Time¶

- Code that solves our graph problems
- Consider which code is simplier
- Consider which code is Faster
22