
From Last Time¶
AminoAcid = {
'A': 'Alanine', 'C': 'Cysteine', 'D': 'Aspartic acid', 'E': 'Glutamic acid',
'F': 'Phenylalanine', 'G': 'Glycine', 'H': 'Histidine', 'I': 'Isoleucine',
'K': 'Lysine', 'L': 'Leucine', 'M': 'Methionine', 'N': 'Asparagine',
'P': 'Proline', 'Q': 'Glutamine', 'R': 'Arginine', 'S': 'Serine',
'T': 'Theronine', 'V': 'Valine', 'W': 'Tryptophan', 'Y': 'Tyrosine',
'*': 'STOP'
}
AminoAbbrv = {
'A': 'Ala', 'C': 'Cys', 'D': 'Asp', 'E': 'Glu',
'F': 'Phe', 'G': 'Gly', 'H': 'His', 'I': 'Ile',
'K': 'Lys', 'L': 'Leu', 'M': 'Met', 'N': 'Asn',
'P': 'Pro', 'Q': 'Gln', 'R': 'Arg', 'S': 'Ser',
'T': 'Thr', 'V': 'Val', 'W': 'Trp', 'Y': 'Tyr',
'*': 'STP'
}
# Now it's time to use this dictionary!
Daltons = {
'A': 71, 'C': 103, 'D': 115, 'E': 129,
'F': 147, 'G': 57, 'H': 137, 'I': 113,
'K': 128, 'L': 113, 'M': 131, 'N': 114,
'P': 97, 'Q': 128, 'R': 156, 'S': 87,
'T': 101, 'V': 99, 'W': 186, 'Y': 163
}
import itertools
def TheoreticalSpectrum(peptide):
# Generate every possible fragment of a peptide
spectrum = set()
for fragLength in xrange(1,len(peptide)+1):
for start in xrange(0,len(peptide)-fragLength+1):
seq = peptide[start:start+fragLength]
spectrum.add(sum([Daltons[res] for res in seq]))
return sorted(spectrum)
def LeaderboardFindPeptide(noisySpectrum, cutThreshold=0.05):
# Golf Tournament Heuristic
spectrum = set(noisySpectrum)
target = max(noisySpectrum)
players = [''.join(peptide) for peptide in itertools.product(Daltons.keys(), repeat=2)]
round = 1
currentLeader = [0.0, '']
while True:
print "%8d Players in round %d [%5.4f]" % (len(players), round, currentLeader[0])
leaderboard = []
for prefix in players:
testSpectrum = set(TheoreticalSpectrum(prefix))
totalWeight = max(testSpectrum)
score = len(spectrum & testSpectrum)/float(len(spectrum | testSpectrum))
if (score > currentLeader[0]):
currentLeader = [score, prefix]
elif (score == currentLeader[0]):
currentLeader += [prefix]
if (totalWeight < target):
leaderboard.append((score, prefix))
remaining = len(leaderboard)
if (remaining == 0):
print "Done, no sequences can be extended"
break
leaderboard.sort(reverse=True)
# Prune the larger of the top 5% or the top 5 players
cut = leaderboard[max(min(5,remaining-1),int(remaining*cutThreshold))][0]
players = [p+r for s, p in leaderboard if s >= cut for r in Daltons.iterkeys()]
round += 1
return currentLeader
2
Let's try a Noisier Spectrum¶
# generate a synthetic experimental spectrum with 60% Error
import random
random.seed(1961)
TyrocidineB1 = "VKLFPWFNQY"
print TyrocidineB1
spectrum = TheoreticalSpectrum(TyrocidineB1)
print len(spectrum)
print spectrum
print
# Pick around ~60% at random to remove
missingMass = random.sample(spectrum[:-1], 30)
print "Missing Masses = ", missingMass
# Add back another ~10% of false, but actual, peptide masses
falseMass = []
for i in xrange(5):
fragment = ''.join(random.sample(Daltons.keys(), random.randint(2,len(TyrocidineB1)-2)))
weight = sum([Daltons[residue] for residue in fragment])
falseMass.append(weight)
print "False Masses = ", falseMass
experimentalSpectrum = sorted(set([mass for mass in spectrum if mass not in missingMass] + falseMass))
print len(experimentalSpectrum)
print experimentalSpectrum
spectrum = TheoreticalSpectrum(TyrocidineB1)
experimentalSpectrum = [mass for mass in spectrum if mass not in missingMass] + falseMass
%time winners = LeaderboardFindPeptide(experimentalSpectrum)
print winners
print len(winners) - 1, "Candidate residues with", winners[0], 'matches'
print TyrocidineB1
print TyrocidineB1 in winners
3
A New Idea¶
- Maybe we are still not using our spectrum to its fullest extent
- Is there some information about missing masses that we can extract?

4
Information in the Mass Differences¶
- Recall the theoretical spectrum of "PLAY" is [71, 97, 113, 163, 184, 210, 234, 281, 347, 444]
- Suppose we remove masses 71 and 163, can we get them back?
- Let's generate a table of all pair-wise differences between the observed peaks
- Notice that interesting numbers, (71, 97, 113, 137, 163, 234) are repeated in the table
| 97 | 113 | 184 | 210 | 234 | 281 | 347 | 444 | |
|---|---|---|---|---|---|---|---|---|
| 97 | 16 | 87 | 113 | 137 | 184 | 250 | 347 | |
| 113 | 71 | 97 | 121 | 168 | 234 | 331 | ||
| 184 | 26 | 50 | 97 | 163 | 260 | |||
| 210 | 24 | 71 | 137 | 234 | ||||
| 234 | 47 | 113 | 210 | |||||
| 281 | 66 | 163 | ||||||
| 347 | 97 |
- Why does this work?
- This table of differences is called the Spectral Convolution
5
Spectral Convolution¶
- Spectral Convolution gives us an approach for recovering some missing masses
- Given a noisy experimental spectrum
- Compute its spectral convolution
- Add frequent masses above some threshold to the spectrum
- Infer the peptide sequence
def SpectralConvolution(spectrum):
delta = {}
for i in xrange(len(spectrum)-1):
for j in xrange(i+1,len(spectrum)):
diff = abs(spectrum[j] - spectrum[i])
delta[diff] = delta.get(diff, 0) + 1
return delta
experimentalSpectrum = sorted(set([mass for mass in spectrum if mass not in missingMass] + falseMass))
specConv = SpectralConvolution(sorted(experimentalSpectrum))
for delta, count in sorted(specConv.iteritems()):
if (count >= 2) and (delta not in experimentalSpectrum) and (delta > min(Daltons.values())):
print "Adding", delta, "appeared", count, "times", "*" if delta in missingMass else ""
experimentalSpectrum.append(delta)
print sorted(missingMass)
winners = LeaderboardFindPeptide(experimentalSpectrum)
print winners
print len(winners) - 1, "Candidate residues with", winners[0], 'matches'
print TyrocidineB1
print TyrocidineB1 in winners
6
A More Realistic Example¶
For long sequences the exponential foundation underlying our heuristic search becomes more evident.
Note: If we set the cutThreshold to 0.0, we will set our cut at the 4th best score and ties with it
Insulin = "MALWMRLLPLLALLALWGPDPAAAFVNQHLCGSHLVEALYLVCGERGFFYTPKTRREAEDLQGSLQPLALEGSLQKRGIVEQCCTSICSLYQLENYCN"
spectrum = TheoreticalSpectrum(Insulin)
print len(spectrum)
missingMass = random.sample(spectrum[:-1], 300)
experimentalSpectrum = sorted([mass for mass in spectrum if mass not in missingMass])
print len(experimentalSpectrum)
# See slide for the following *hack*
del Daltons['I']
del Daltons['K']
%time winners = LeaderboardFindPeptide(experimentalSpectrum, cutThreshold=0.01)
print winners
print len(winners) - 1, "Candidate residues with", winners[0], 'matches'
print Insulin
print Insulin in winners
# See slide for the following *hack*
Daltons['I'] = Daltons['L']
Daltons['K'] = Daltons['Q']
Some of the reasons that things blow up:
- The search space got large fast
- There must be a LOT of ties
- Algorithm tends to keep all (N-k+1) subpeptides as k approaches the sequence's size (k is related to our round)
- The I/L and K/Q ambiguities lead to exponential number of ties, hence the hack
- Reversed sequences are doubling our leaderboard size
There are bandaids to fix problems 3 and 4, but the problem remains
7
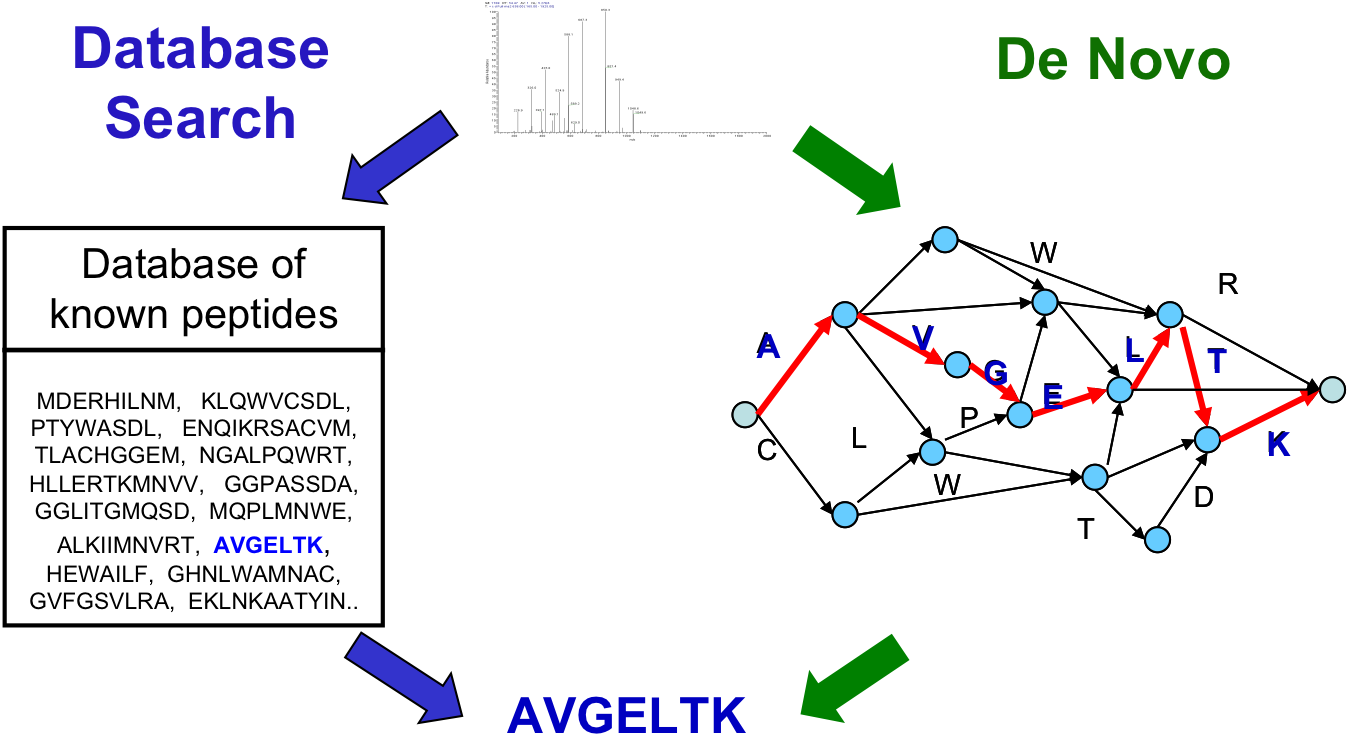
Peptide Identification Problem¶
Goal: Find a peptide from the database with maximal match between an experimental and theoretical spectrum.
Input:
- S: experimental spectrum
- database of peptides
- Δ: set of possible ion types
- m: parent mass
Output:
- A peptide of mass m from the database whose theoretical spectrum matches the experimental S spectrum the best
9
Mass Spec Database Searches¶
How do you get a database?
- Compute theoretical spectrums for all peptides from length N to M
- More commonly, store theoretical spectrums for known peptide sequences
Database searches are very effective in identfying known or closely related proteins.
Experimental spectrums are compared with spectra of database peptides to find the best fit (ex. SEQUEST, Yates et al., 1995)
But reliable algorithms for identification of new proteins is a more difficult problem.
10
Essence of the Database Search¶
We need a notion of spectral similarity that correlates well with the sequence similarity.
If peptides are a few mutations/modifications apart, the spectral similarity between their spectra should be high.
Simplest measure: Shared Peak Counts (SPC)
- Very similar to the scoring function used in our De novo approach.
11

print TheoreticalSpectrum('PRTEIN')
print TheoreticalSpectrum('PRTEYN')
print TheoreticalSpectrum('PGTEYN')
print set(TheoreticalSpectrum('PRTEIN')) & set(TheoreticalSpectrum('PRTEYN'))
print set(TheoreticalSpectrum('PRTEIN')) & set(TheoreticalSpectrum('PGTEYN'))
Spectral Convolution to the Rescue!¶
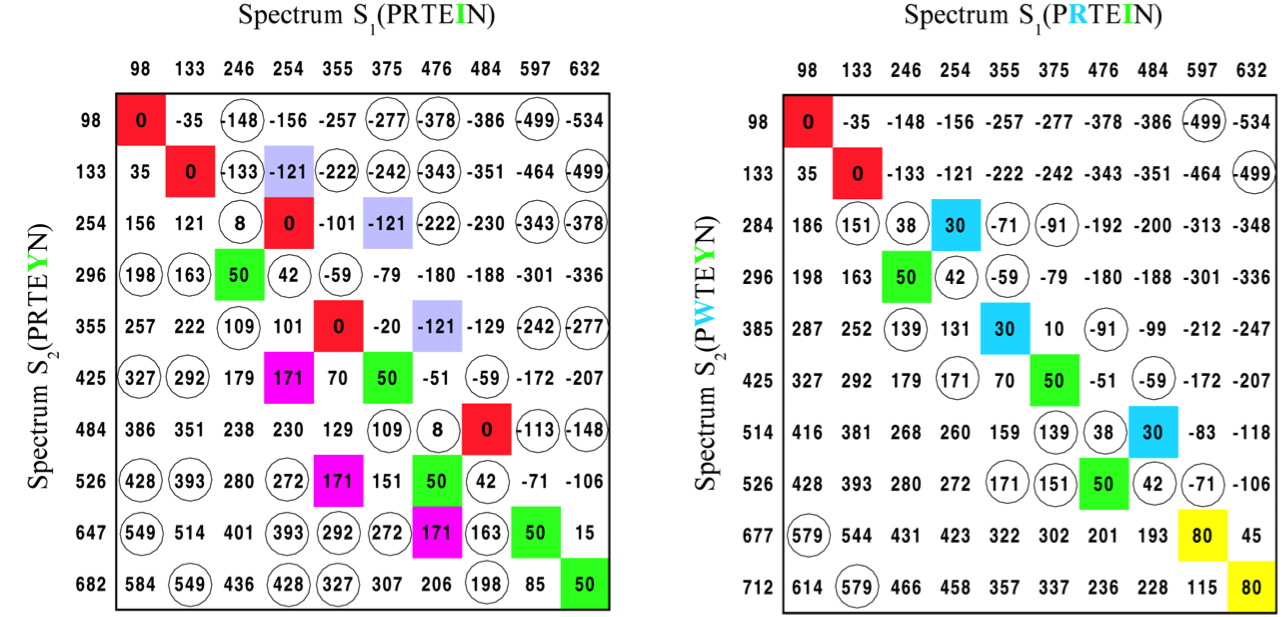
Difference matrix of spectrums. The elements with multiplicity >2 are colored; the elements with multiplicity =2 are circled. The SPC takes into account only the red entries
12