Determining a Peptide's Sequence¶
|

|
1
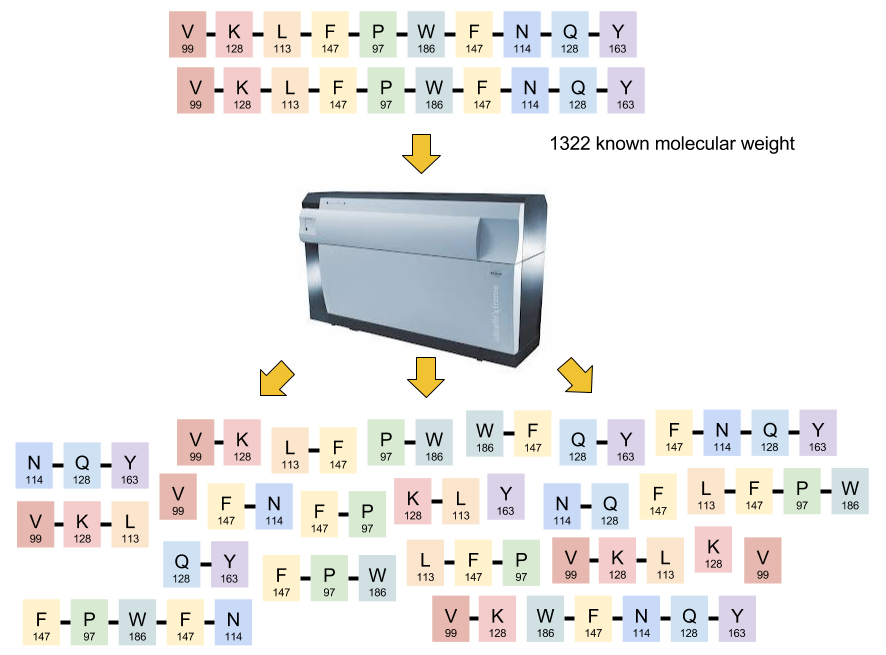
Structure of a Peptide Chain¶
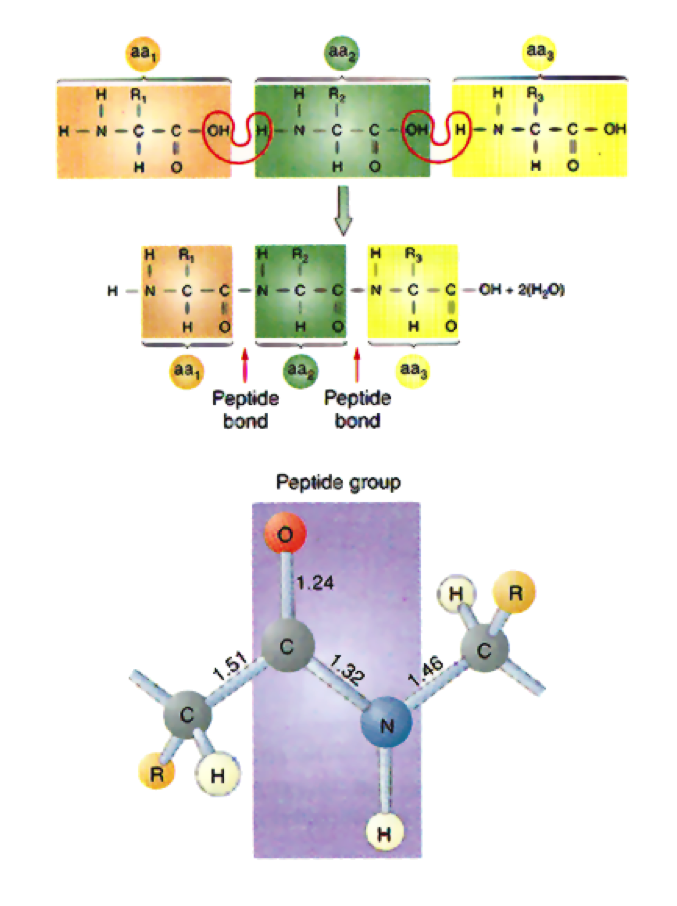
- Peptides are chains of amino acids that are joined by peptide bonds
- These bonds reduce the weight of each amino acid by one H20 molecule
- The result is called a residue
- A Mass Spectrograph can precisely measure the molecular weight (and charge and abundance) of any peptide chain
- Since the molecular weight of each of the possible 20 residues is known precisely, one can ask the question, which combination of residues would give a particular weight?
- The problem is ambiguous for the entire molecule
- Consider all permulations of 'PIT':
'PIT', 'PTI', 'ITP', 'IPT', 'TPI', and 'TIP' all weigh the same - But they differ in their 2-peptide fragments:
'PIT' breaks into 'PI' and 'IT', while 'PTI' breaks into 'PT' and 'TI'
- Consider all permulations of 'PIT':
3
An Simplified Peptide Weight table¶
- The actual molecular weight of an amino acid is a real number. This acounts for the relative abundances of atomic isotopes
- We will use a simplified version that uses only integer molecular weights
Example:
Molecular weight of Glycine Amino Acid
$$W(C_2 H_5 N O_2) = 12 \times 2 + 5 \times 1 + 14 + 16 \times 2 = 75$$
Molecular wieght of Glycine Residue (Minus the $H_{2} O$ lost forming the peptide bond)
$$W(C_2 H_5 N O_2 - H_2 O) = 57$$
- We can repeat this for all 20 Amino Acids to get a integer molecular weight table, which I call Daltons
4
Table Definitions¶
AminoAcid = {
'A': 'Alanine', 'C': 'Cysteine', 'D': 'Aspartic acid', 'E': 'Glutamic acid',
'F': 'Phenylalanine', 'G': 'Glycine', 'H': 'Histidine', 'I': 'Isoleucine',
'K': 'Lysine', 'L': 'Leucine', 'M': 'Methionine', 'N': 'Asparagine',
'P': 'Proline', 'Q': 'Glutamine', 'R': 'Arginine', 'S': 'Serine',
'T': 'Theronine', 'V': 'Valine', 'W': 'Tryptophan', 'Y': 'Tyrosine',
'*': 'STOP'
}
AminoAbbrv = {
'A': 'Ala', 'C': 'Cys', 'D': 'Asp', 'E': 'Glu',
'F': 'Phe', 'G': 'Gly', 'H': 'His', 'I': 'Ile',
'K': 'Lys', 'L': 'Leu', 'M': 'Met', 'N': 'Asn',
'P': 'Pro', 'Q': 'Gln', 'R': 'Arg', 'S': 'Ser',
'T': 'Thr', 'V': 'Val', 'W': 'Trp', 'Y': 'Tyr',
'*': 'STP'
}
# Now it's time to use this dictionary!
Daltons = {
'A': 71, 'C': 103, 'D': 115, 'E': 129,
'F': 147, 'G': 57, 'H': 137, 'I': 113,
'K': 128, 'L': 113, 'M': 131, 'N': 114,
'P': 97, 'Q': 128, 'R': 156, 'S': 87,
'T': 101, 'V': 99, 'W': 186, 'Y': 163
}
5
Some Issues with our Table¶

- We can't distinguish between Leucine (L) and Isoleucine (I). They both weight 113 d
- Nor can we distinguish Lysine (K) and Glutamine (Q), which weigh 128 d
- For long peptide chains >50, our errors can build up
- In reality, peptides can loose or gain one or more small molecules from their side chains and fractured peptide bonds
- Hydrogen ions (H - 1 Dalton)
- Water (H2O - 18 Daltons)
- Ammonia (NH3, 17 Daltons)
- This leads to measurements that vary around the ideal sums we assume
- Regardless of these caveats, let's keep going
6
Let's compute the Total Molecular Weight of our Target¶
TyrocidineB1 = "VKLFPWFNQY"
# The weight of Tyrocidine B1
print sum([Daltons[res] for res in TyrocidineB1])
- Generally, we will assume that this target weight is known
- We will use it as a terminating condition for many of our algorithms that attempt to reconstruct the measured set of weights
7
Ideally, what Weights should we get?¶
- We will make the optimistic assumption that we will fracture our given petide chain into all of its constituent parts
- For a 10 peptide chain
- 10 single peptides
- 9, 2-peptide chains
- 8, 3-peptide chains
- 7, 4-peptide chains
- 6, 5-peptide chains
- 5, 6-peptide chains
- 4, 7-peptide chains
- 3, 8-peptide chains
- 2, 9-peptide chains
- 1, 10-peptide chain
- This gives an upper bound of ${10 \choose 2} = 55$ molecular weights, but relatity both the peptide chains and their weights may not be unique
- The collection of all possible sub-peptide molecular weights from a peptide is called the peptide's Theoretical Spectrum
8
Code for computing a Theoretical Spectrum¶
def TheoreticalSpectrum(peptide):
# Generate every possible fragment of a peptide
spectrum = set()
for fragLength in xrange(1,len(peptide)+1):
for start in xrange(0,len(peptide)-fragLength+1):
seq = peptide[start:start+fragLength]
spectrum.add(sum([Daltons[res] for res in seq]))
return sorted(spectrum)
print TyrocidineB1
spectrum = TheoreticalSpectrum(TyrocidineB1)
print len(spectrum)
print spectrum
- Why are we using a set rather than a list? Notice that we end up returning a list.
peptide = TyrocidineB1
fragList = []
for fragLength in xrange(1,len(peptide)+1):
for start in xrange(0,len(peptide)-fragLength+1):
seq = peptide[start:start+fragLength]
fragList.append((sum([Daltons[res] for res in seq]), seq))
print len(fragList)
lastWeight = 0
for weight, frag in sorted(fragList):
print "%10s: %d%s" % (frag, weight, "*" if (weight == lastWeight) else " ")
lastWeight = weight
- Here's a smaller example that we will play with first before solving for Tyrocidine B1
spectrum = TheoreticalSpectrum('PLAY')
print len(spectrum)
print spectrum
9
Can we Invert the Process of creating a Spectrum?¶

- In essence, the problem of inferring a peptide chain from the set of mass values reported by a Mass Spectrometer is the inverse of the code we just wrote
Hard Problem: Peptide Sequence ← Spectrum
- Why is computing a spectrum from a peptide sequence easy? $O(N^2)$?
- Why is computing a peptide sequence from a specturm hard? $O(?)$
10
How might you approach this problem?¶

Can you think of a Brute-Force way of solving this problem?
Here's one:
- For every peptide sequence with the target peptide's molecular weight
- Compute the sequence's Theoretical Spectrum
- If it matches the one given, report this peptide as a possible solution
- Which step in this algorithm is the hard part?
11
A Brute-Force Attempt¶
def PossiblePeptide(spectrum, prefix=''):
""" A brute force method of generating all peptide
sequences that add up to our target weight
from the given spectrum """
global peptideList
if (len(prefix) == 0):
peptideList = []
current = sum([Daltons[res] for res in prefix])
target = max(spectrum) # our target
if (current == target):
peptideList.append(prefix)
elif (current < target):
for residue in Daltons.iterkeys():
PossiblePeptide(spectrum, prefix+residue)
def TestPeptides(candidateList, target):
filteredList = []
for peptide in candidateList:
candidateSpectrum = TheoreticalSpectrum(peptide)
if (candidateSpectrum == target):
filteredList.append(peptide)
return filteredList
spectrum = TheoreticalSpectrum('PLAY')
%time PossiblePeptide(spectrum)
print len(peptideList), "candidates"
print "PLAY" in peptideList
%time matches = TestPeptides(peptideList, spectrum)
print matches
print "PLAY" in matches
12
Impressions?¶
- Not so bad for a first attempt, but how will it perform for longer peptides?
- We are getting the expected answer as well as answers with the indistinguishable amino acids substituted
- We are also getting the sequence reversed? Is this a surprise?
- We could code around this, but for today we'll just include the reversed peptide chain as a possible answer
13
Improving on Brute Force¶
The brute force method does not make good use of the spectrum it is given
- It only ever considers the largest value from this table
- How might we make use of the other values?
We could only extend our prefix with residues that appear in our spectrum
- The weight of every new prefix that we consider should also be in our spectrum
Actual fragments:
P L A Y PL LA AY PLA LAY PLAY
Growing and Checking prefixes:
A I L P Y
AI = LA IA = LA LA = LA PI = PL YA = AY
AIP = PLA IAP = PLA LAP = PLA PIA = PLA YAI = LAY
AIPY = PLAY IAPY = PLAY LAPY = PLAY PIAY = PLAY YAIP = PLAY
AIY = LAY IAY = LAY LAY = LAY YAL = LAY
AIYP = PLAY IAYP = PLAY LAYP = PLAY YALP = PLAY
AL = LA IP = PL LP = PL PL = PL
ALP = PLA IPA = PLA LPA = PLA PLA = PLA
ALPY = PLAY IPAY = PLAY LPAY = PLAY PLAY = PLAY
ALY = LAY
ALYP = PLAY
AY = AY
AYI = LAY
AYIP = PLAY
AYL = LAY
AYLP = PLAY
14
Only a Small Change to the Code¶
def ImprovedPossiblePeptide(spectrum, prefix=''):
global peptideList
if (len(prefix) == 0):
peptideList = []
current = sum([Daltons[res] for res in prefix])
target = max(spectrum)
if (current == target):
peptideList.append(prefix)
elif (current < target):
for residue in Daltons.iterkeys():
if (Daltons[residue] not in spectrum):
continue
extend = prefix + residue
if (sum([Daltons[res] for res in extend]) not in spectrum):
continue
ImprovedPossiblePeptide(spectrum, extend)
spectrum = TheoreticalSpectrum('PLAY')
%time ImprovedPossiblePeptide(spectrum)
print len(peptideList)
print "PLAY" in peptideList
%time matches = TestPeptides(peptideList, spectrum)
print matches
print "PLAY" in matches
- Provides a HUGE performace difference
- Yet another example of Branch-and-Bound
- We improved both the enumeration and verification phases, but the differece was much more significant in the enumeration step
- Let's look at the list generated by pruning the search tree and see if it gives us any more insights
for peptide in peptideList:
print peptide
TheoreticalSpectrum('PLAY')
TheoreticalSpectrum('LAPY')
print sum([Daltons[res] for res in 'AP']) # Suffix of 'LAP' prefix
print sum([Daltons[res] for res in 'APY']) # Suffix of 'LAPY'
print sum([Daltons[res] for res in 'PY']) # Suffix of 'LAPY'
- There are still differences in the spectrums, yet every prefix was in the spectrum when we added it. What are we missing?
- Suffixes!
15
We can do Even Better¶
- All suffixes of each prefix that we consider should also be in our spectrum
def UltimatePossiblePeptide(spectrum, prefix=''):
global peptideList
if (len(prefix) == 0):
peptideList = []
current = sum([Daltons[res] for res in prefix])
target = max(spectrum)
if (current == target):
peptideList.append(prefix)
elif (current < target):
for residue in Daltons.iterkeys():
extend = prefix + residue
suffix = [extend[i:] for i in xrange(len(extend))]
for fragment in suffix:
if (sum([Daltons[res] for res in fragment]) not in spectrum):
break
else:
UltimatePossiblePeptide(spectrum, extend)
spectrum = TheoreticalSpectrum('PLAY')
%time UltimatePossiblePeptide(spectrum)
print len(peptideList)
print "PLAY" in peptideList
%time matches = TestPeptides(peptideList, spectrum)
print matches
print "PLAY" in matches
- A little slower, but our list is pruned significantly
- All of theses have identical spectrums
16
Now let's return to our real peptide¶
spectrum = TheoreticalSpectrum(TyrocidineB1)
%time UltimatePossiblePeptide(spectrum)
print len(peptideList)
print TyrocidineB1 in peptideList
%time matches = TestPeptides(peptideList, spectrum)
print len(matches)
print TyrocidineB1 in matches
for peptide in peptideList:
print peptide
All of these peptides give also give us our desired spectrum
17
Great, but our assumptions are a little Naïve
- In relaity, Mass Spectometers don't report the Theoretical Spectrum of a peptide
- Instead they report a measured or Experimental Spectrum
- This spectrum might miss some fragments
- It might also report false fragments
- From Contaminants
- New peptides forming from assorted fragments
- The result is that some of the masses that appear may be misleading, and some that we want might be missing
- We need to make our algorithms for reporting candidate protein sequences robust to noise
18
An example Experimental Spectrum for Tyrocidine B1¶
97, 99, 113, 114, 128, 147, 163,
186, 200, 227, 241, 242, 244, 260,
261, 283, 291, 333, 340, 357, 388,
389, 405, 430, 447, 457, 485, 487,
543, 544, 552, 575, 577, 584, 659,
671, 672, 690, 691, 731, 738, 770,
804, 818, 819, 835, 906, 917, 932,
982, 1031, 1060, 1095, 1159, 1223, 1322
False Masses: present in the experimental spectrum, but not in the theoretical spectrum
Missing Masses: present in the theoretical spectrum, but not in the experimental spectrum
19
An example Experimental Spectrum for Tyrocidine B1¶
97, 99, 113, 128, 147, 163,
186, 200, 227, 241, 242, 244, 260,
261, 283, 291, 333, 340, 357,
405, 430, 447, 457, 487,
543, 544, 552, 575, 577, 584, 659,
671, 672, 690, 691, 731, 738, 770,
804, 818, 819, 835, 906, 917, 932,
982, 1031, 1095, 1159, 1322
False Masses: We don't know which these are
Missing Masses: And these values don't appear
20
An aside: How I faked an Experimental Spectrum¶
# generate a synthetic experimental spectrum with 10% Error
import random
random.seed(1961)
spectrum = TheoreticalSpectrum(TyrocidineB1)
# Pick around ~10% at random to remove
missingMass = random.sample(spectrum, 6)
print "Missing Masses = ", missingMass
# Add back another ~10% of false, but actual, peptide masses
falseMass = []
for i in xrange(5):
fragment = ''.join(random.sample(Daltons.keys(), random.randint(2,len(TyrocidineB1)-2)))
weight = sum([Daltons[residue] for residue in fragment])
falseMass.append(weight)
print "False Masses = ", falseMass
experimentalSpectrum = sorted(set([mass for mass in spectrum if mass not in missingMass] + falseMass))
print experimentalSpectrum
21
A Golf Tournament Analogy¶
- After the first couple of rounds of a major golf tournament a cut is made of all golfers who are so far back from the leader that it is deemed they are unlikely to ever finish in the money
- These cut golfers are removed from further consideration
- This choice is heuristic
- It is possible that a player just below the cut could have two exceptional rounds, but that is considered unlikely
- What is the equivalent of a score in our peptide finding problem?
- The number of matching masses in the candidate peptide's Theoretical Spectrum and the Experimental Spectrum
- Normalized score, why?
- matches / len(candidate peptide's Theoretical Spectrum)
- In our peptide golf game a round will be considered a one peptide extension of a active set of player peptides
- We will do cuts on every round, keeping to top 5% of finishers or the top 5 players, which ever is more
- Why 5%? It is arbitrary, but on each round we will extend the current set of players by one of 20 amino acids, thus increasing the number of peptides by a factor of 20, so reducing by 5% leaves the poolsize realtively stable.
22
An Implementation¶
import itertools
def LeaderboardFindPeptide(noisySpectrum, cutThreshold=0.05):
# Golf Tournament Heuristic
spectrum = set(noisySpectrum)
target = max(noisySpectrum)
players = [''.join(peptide) for peptide in itertools.product(Daltons.keys(), repeat=2)]
round = 1
currentLeader = [0.0, '']
while True:
print "%8d Players in round %d" % (len(players), round)
leaderboard = []
for prefix in players:
testSpectrum = set(TheoreticalSpectrum(prefix))
totalWeight = max(testSpectrum)
score = len(spectrum & testSpectrum)/float(len(testSpectrum))
if (totalWeight == target):
if (score > currentLeader[0]):
currentLeader = [score, prefix]
elif (score == currentLeader[0]):
currentLeader += [prefix]
elif (totalWeight < target):
leaderboard.append((score, prefix))
remaining = len(leaderboard)
if (remaining == 0):
print "Done, no sequences can be extended"
break
leaderboard.sort(reverse=True)
# Prune the larger of the top 5% or the top 5 players
cut = leaderboard[max(min(5,remaining-1),int(remaining*cutThreshold))][0]
players = [p+r for s, p in leaderboard if s >= cut for r in Daltons.iterkeys()]
round += 1
return currentLeader
spectrum = TheoreticalSpectrum(TyrocidineB1)
experimentalSpectrum = [mass for mass in spectrum if mass not in missingMass] + falseMass
%time winners = LeaderboardFindPeptide(experimentalSpectrum)
print winners
print len(winners) - 1, "Candidate residues with", winners[0], 'matches'
print TyrocidineB1
print TyrocidineB1 in winners
23
Next Time¶
- This method works well, but it rely on heuristcs, and thus might miss the best answer
- Our methods are still make a lot of simplfying assumptions
- In reality relying only exact matches might lead us
- We will continue to explore ways of assembling peptide sequences from a given experimental spectrum
24