Finding TFBS Motifs in our Lifetime¶

- Recall from last time that the Brute Force approach for finding a common 10-mer motif common to 10 sequences of length 80 bases was going to take up roughly 30,000 years
- Today well consider alternative and non-obvious approaches for solving this problem
- We will trade one old man (us) for another (an Oracle)
Note: Our Python/Jupyter study session will be held in SN014 on 1/28 from 5:00pm to 6:30pm
1
Recall from last Lecture¶
- The following set of 10 sequences have an embedded noisy motif, TAGATCCGAA.
1 tagtggtcttttgagtgTAGATCTGAAgggaaagtatttccaccagttcggggtcacccagcagggcagggtgacttaat
2 cgcgactcggcgctcacagttatcgcacgtttagaccaaaacggagtTGGATCCGAAactggagtttaatcggagtcctt
3 gttacttgtgagcctggtTAGACCCGAAatataattgttggctgcatagcggagctgacatacgagtaggggaaatgcgt
4 aacatcaggctttgattaaacaatttaagcacgTAAATCCGAAttgacctgatgacaatacggaacatgccggctccggg
5 accaccggataggctgcttatTAGGTCCAAAaggtagtatcgtaataatggctcagccatgtcaatgtgcggcattccac
6 TAGATTCGAAtcgatcgtgtttctccctctgtgggttaacgaggggtccgaccttgctcgcatgtgccgaacttgtaccc
7 gaaatggttcggtgcgatatcaggccgttctcttaacttggcggtgCAGATCCGAAcgtctctggaggggtcgtgcgcta
8 atgtatactagacattctaacgctcgcttattggcggagaccatttgctccactacaagaggctactgtgTAGATCCGTA
9 ttcttacacccttcttTAGATCCAAAcctgttggcgccatcttcttttcgagtccttgtacctccatttgctctgatgac
10 ctacctatgtaaaacaacatctactaacgtagtccggtctttcctgatctgccctaacctacaggTCGATCCGAAattcg
2
Consensus Scoring Function¶
- We developed a consensus scoring function to overcome the noise, but we needed to apply it an exponential number, $O(N^t)$ of times!
- Here's the scoring function...
def Score(s, DNA, k):
"""
compute the consensus SCORE of a given k-mer
alignment given offsets into each DNA string.
s = list of starting indices, 1-based, 0 means ignore
DNA = list of nucleotide strings
k = Target Motif length
"""
score = 0
for i in xrange(k):
# loop over string positions
cnt = dict(zip("acgt",(0,0,0,0)))
for j, sval in enumerate(s):
# loop over DNA strands
base = DNA[j][sval+i]
cnt[base] += 1
score += max(cnt.itervalues())
return score
3
And here's the Score we're looking for...¶
seqApprox = [
'tagtggtcttttgagtgtagatctgaagggaaagtatttccaccagttcggggtcacccagcagggcagggtgacttaat',
'cgcgactcggcgctcacagttatcgcacgtttagaccaaaacggagttggatccgaaactggagtttaatcggagtcctt',
'gttacttgtgagcctggttagacccgaaatataattgttggctgcatagcggagctgacatacgagtaggggaaatgcgt',
'aacatcaggctttgattaaacaatttaagcacgtaaatccgaattgacctgatgacaatacggaacatgccggctccggg',
'accaccggataggctgcttattaggtccaaaaggtagtatcgtaataatggctcagccatgtcaatgtgcggcattccac',
'tagattcgaatcgatcgtgtttctccctctgtgggttaacgaggggtccgaccttgctcgcatgtgccgaacttgtaccc',
'gaaatggttcggtgcgatatcaggccgttctcttaacttggcggtgcagatccgaacgtctctggaggggtcgtgcgcta',
'atgtatactagacattctaacgctcgcttattggcggagaccatttgctccactacaagaggctactgtgtagatccgta',
'ttcttacacccttctttagatccaaacctgttggcgccatcttcttttcgagtccttgtacctccatttgctctgatgac',
'ctacctatgtaaaacaacatctactaacgtagtccggtctttcctgatctgccctaacctacaggtcgatccgaaattcg']
print Score([17, 47, 18, 33, 21, 0, 46, 70, 16, 65], seqApprox, 10)
%timeit Score([17, 47, 18, 33, 21, 0, 46, 70, 16, 65], seqApprox, 10)
So even at a blazing $50.6 \mu s$ we'll need many lifetimes to compute the $70^{10}$ scores
4
Pruning Trees¶

- One method for reducing the computational cost of a search algorithm is to prune the space of permutations that could not possibly lead to a better answer than the current best answer.
- Pruning decisions are based on solutions to subproblems that appear early on and offer no hope
- How does this apply to our Motif finding problem?
Consider any permutation of offsets that begins with the indices [25, 63, 10, 43, ....]. Just based on the first 4 indices the largest possible score is 17 + 6*10 = 87, which assumes that all 6 remaining strings match perfectly at all 10 positions.
DNA[0][25:35] a a g g g a a a g t
DNA[1][63:73] g t t t a a t c g g
DNA[2][10:20] a g c c t g g t t a
DNA[3][43:53] t t g a c c t g a t
a [2, 1, 0, 1, 1, 2, 1, 1, 1, 1]
Profile c [0, 0, 1, 1, 1, 1, 0, 1, 0, 0]
g [1, 1, 2, 1, 1, 1, 1, 1, 2, 1]
t [1, 2, 1, 1, 1, 0, 2, 1, 1, 2]
[2, 2, 2, 1, 1, 2, 2, 1, 2, 2] Score = 17
If the best answer so far is 89, there is no need to consider the 706 offset permuations that start with these 4 indices.
5
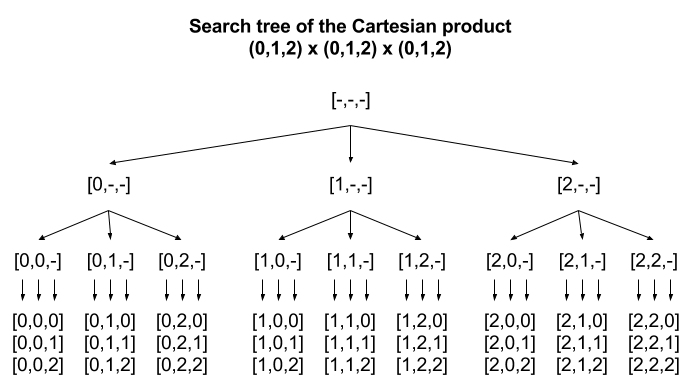
Search Trees¶
- Our standard method for enumerating permutations can be considered as a traversal of leaf nodes in a search tree
- Suppose after checking the first few offsets could know already that any score of children nodes could not beat the best score seen so far?
6
Branch-and-Bound Motif Search¶
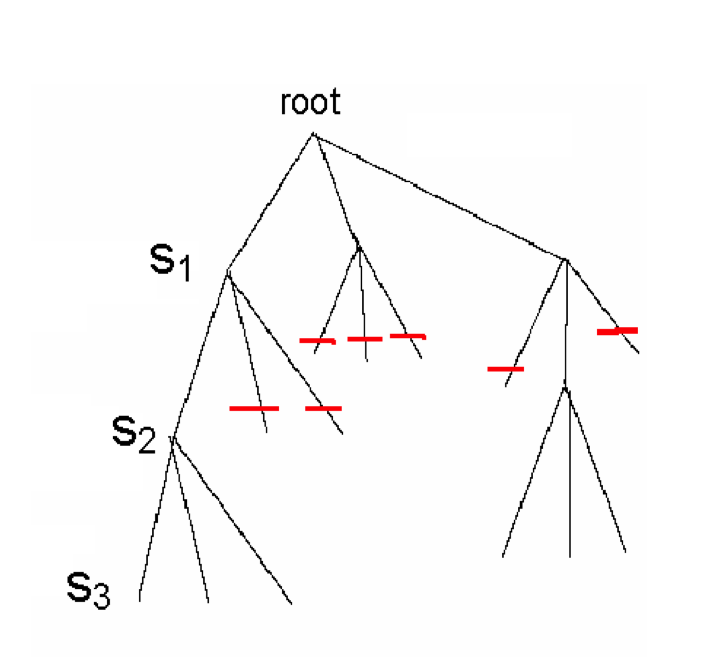
- Since each level of the tree goes deeper into search, discarding a prefix discards all following branches
- This saves us from looking at $(N – k + 1)^{t-depth}$ leaves
- Note our enumeration of tree-branches is depth-first
- We'll formulate of trimming algorithm as a recursive algorithm
5
A Recursive Exploration of a Search Tree¶
bestAlignment = []
prunedPaths = 0
def exploreMotifs(DNA,k,path,bestScore):
""" Recursively finds a k-length motif in the
list of DNA sequences by exploring all paths
in a search tree. Each recursive call extends
path by one index, once the path reaches the
number of DNA strings a score is computed. """
global bestAlignment, prunedPaths
depth = len(path)
t = len(DNA)
if (depth == t):
s = Score(path,DNA,k)
if (s > bestScore):
bestAlignment = [p for p in path]
return s
else:
return bestScore
else:
# Before going down this path further let's
# consider if an optimistic best score can
# possibly beat the best score so far
if (depth > 1):
OptimisticScore = k*(t-depth) + Score(path,DNA,k)
else:
OptimisticScore = k*t
if (OptimisticScore < bestScore):
prunedPaths = prunedPaths + 1
return bestScore
else:
for s in xrange(len(DNA[depth])-k+1):
newPath = tuple([i for i in path] + [s])
bestScore = exploreMotifs(DNA,k,newPath,bestScore)
return bestScore
def BranchAndBoundMotifSearch(DNA, k):
""" Finds a k-length motif within a list of DNA sequences"""
global bestAlignment, prunedPaths
bestAlignment = []
prunedPaths = 0
bestScore = 0
for s in xrange(len(DNA[0])-k+1):
bestScore = exploreMotifs(DNA,k,[s],bestScore)
print bestAlignment, bestScore, prunedPaths
%time BranchAndBoundMotifSearch(seqApprox[0:6], 10)
7
Observations¶

- For our problem instance, Branch-and-Bound Motif finding is significantly faster
- It found a motif in the first 6 strings in less time than the Brute Force approach found a solution in the first 4 strings
- More than $70^2 \approx 5000$ times faster
- It did so by trimming more than 8 Million paths
- Trimming added extra calls to Score (basically doubling the worst-case number of calls), but ended up saving even more hopeless calls along longer paths.
- In practice, Branch-and-Bound, significantly improved the average performance
- Does this improve the worst-case performance from $O(tkN^t)$?
- What if all of our motifs were found at the end of each DNA string?
- How do we avoid these worse case data sets?
- Randomize the search-tree tranversal order
8
We Need A Fresh New Approach¶

- Enumerating every possible permuation of motif positions is not getting us the speed we want.
- Let's try another tried and tested apprach to algorithm design, mixing up the problem
- Suppose that some Oracle could tell us what the motif is
- How long would it take us to find its position in each string?
- We could compute the Hamming Distance from our given motif to the k-mer at every position of each DNA sequence and keep track of the smallest distance and its position on each string.
- These positions are our best guess of where the motif can be found on each string
- Let's this approach to scanning-and-scoring a given motif.
9
Scanning-and-Scoring a Motif¶
def ScanAndScoreMotif(DNA, motif):
totalDist = 0
bestAlignment = []
k = len(motif)
for seq in DNA:
minHammingDist = k+1
for s in xrange(len(seq)-k+1):
HammingDist = sum([1 for i in xrange(k) if motif[i] != seq[s+i]])
if (HammingDist < minHammingDist):
bestS = s
minHammingDist = HammingDist
bestAlignment.append(bestS)
totalDist += minHammingDist
return bestAlignment, totalDist
print ScanAndScoreMotif(seqApprox, "tagatccgaa")
%timeit ScanAndScoreMotif(seqApprox, "tagatccgaa")
Wow, we can test over 500 motifs per second!
10
Scan-and-Score Motif Performance¶
- There are $t (N - k + 1)$ positions to test the motif, and each test requires $k$ tests.
- So each scan is $O(t N k)$.
- So where where do we get candidate motifs?
- Can we try all of them? There are $4^{10} = 1048576$ in our example.
- Do the math, 1048576 motifs × 2 mS ≈ 35 mins
- Not fast, but not a lifetime
- This approach is called a Median String Motif Search
- Recall from last Lecture that a string that minimizes Hamming distance is like finding a middle or median string that is closer to all instances than the instances are to each other.
11
Let's Do It¶
import itertools
def MedianStringMotifSearch(DNA,k):
""" Consider all possible 4**k motifs"""
bestAlignment = []
minHammingDist = k*len(DNA)
kmer = ''
for pattern in itertools.product('acgt', repeat=k):
motif = ''.join(pattern)
align, dist = ScanAndScoreMotif(DNA, motif)
if (dist < minHammingDist):
bestAlignment = [s for s in align]
minHammingDist = dist
kmer = motif
return bestAlignment, minHammingDist, kmer
%time MedianStringMotifSearch(seqApprox,10)
Should we declare victory and move on? Do you find anything uncomfortable about an algorithm that requires $O(t N k 4^k)$ steps?
12
Notes on Median String Motif Search¶
- There are similarities between finding and alignment with minimal Hamming Distance and maximizing a Motif's consensus score.
- In fact, if instead of counting mismatches as in the code fragment:
HammingDist = sum([1 for i in xrange(k) if motif[i] != seq[s+i]])
we had counted matchesMatches = sum([1 for i in xrange(k) if motif[i] == seq[s+i]])
and found the maximum(TotalMatches) instead of the min(TotalHammingDistance) we would be using the same measure as Score(). - Thus, we expect MedianStringMotifSearch() to give the same answer as either BruteForceMotifSearch() or BranchAndBoundMotifSearch().
- However, the $4^k$ term raises some concerns. If k were instead 20, then we'd have to Scan-and-Score more than $10^12$ times. Another not-in-my-lifetime algorithm
- We can also apply the Branch-and-Bound approach to the Median string method, but, as before it would only improve the average case.
13
Other ways to guess the Motif?¶
- If we knew that the motif that we are looking for was contained somewhere in our DNA sequences we could test the $(N-k+1)t$ motifs from our DNA, giving a $O(N^2t^2)$ algorithm.
- Unfortunately, as you may recall our motif did not appear actually appear in our data.
- You could keep track of a few good motif candidates using a manageable and perhaps random subsets of the given DNA sequences, and use them as your candidate motifs.
14
Let's try considering only Motifs seen in the DNA¶
def ContainedMotifSearch(DNA,k):
""" Consider only motifs from the given DNA sequences"""
motifSet = set()
for seq in DNA:
for i in xrange(len(seq)-k+1):
motifSet.add(seq[i:i+k])
print "%d Motifs in our set" % len(motifSet)
bestAlignment = []
minHammingDist = k*len(DNA)
kmer = ''
for motif in motifSet:
align, dist = ScanAndScoreMotif(DNA, motif)
if (dist < minHammingDist):
bestAlignment = [s for s in align]
minHammingDist = dist
kmer = motif
return bestAlignment, minHammingDist, kmer
%time ContainedMotifSearch(seqApprox,10)
Not exactly the motif we were looking for (off by a 'g'),
[17, 47, 18, 33, 21, 0, 46, 70, 16, 65], 11, 'tagatccgaa'but boy was it fast! Where's a good Oracle when you need one?
16
Perhaps the Consensus Score matrix can provide insight¶
If we call Score([17, 47, 18, 33, 21, 0, 46, 70, 16, 65], seqApprox, 10)
DNA[0][17:27] t a g a t c t g a a
DNA[1][31:41] t a g a c c a a a a
DNA[2][18:28] t a g a c c c g a a
DNA[3][33:43] t a a a t c c g a a
DNA[4][21:31] t a g g t c c a a a
DNA[5][ 0:10] t a g a t t c g a a
DNA[6][46:56] c a g a t c c g a a
DNA[7][70:80] t a g a t c c g t a
DNA[8][16:26] t a g a t c c a a a
DNA[9][65:75] t c g a t c c g a a
a [0, 9, 1, 9, 0, 0, 1, 3, 9,10]
c [1, 1, 0, 0, 2, 9, 8, 0, 0, 0]
g [0, 0, 9, 1, 0, 0, 0, 7, 0, 0]
t [9, 0, 0, 0, 8, 1, 1, 0, 1, 0]
[9, 9, 9, 9, 8, 9, 8, 7, 9,10] Score = 87
Consensus t a g a t c c g a a Our motif!
Any Ideas?
17
Contained Consensus Motif Search¶
def Consensus(s, DNA, k):
"""
compute the consensus k-Motif of an alignment
given offsets into each DNA string.
s = list of starting indices, 1-based, 0 means ignore
DNA = list of nucleotide strings
k = Target Motif length
"""
consensus = ''
for i in xrange(k):
# loop over string positions
cnt = dict(zip("acgt",(0,0,0,0)))
for j, sval in enumerate(s):
# loop over DNA strands
base = DNA[j][sval+i]
cnt[base] += 1
consensus += max(cnt.iteritems(), key=lambda tup: tup[1])[0]
return consensus
def ContainedConsensusMotifSearch(DNA,k):
bestAlignment, minHammingDist, kmer = ContainedMotifSearch(DNA,k)
motif = Consensus(bestAlignment,DNA,k)
newAlignment, HammingDist = ScanAndScoreMotif(DNA, motif)
return newAlignment, HammingDist, motif
%time ContainedConsensusMotifSearch(seqApprox,10)
Now we're cooking!
18
Dad, are we there yet?¶

We got the answer that we were looking for, but
- How can we be sure it will always give the correct answer?
- Our other methods were exhaustive, they examined every possibility
- This method considers only a subset of solutions, picks the best one in a greedy fashion
- What it there had been ties amoung the candidate motifs?
- What if the consensus score (87% matches) had been lower
- Would we, should we, be satisfied?
- It's one thing to be greedy, and another to be both greedy and biased
- Our method is greedy in that it considers only the best contained motif, greedy methods are subject to falling into local minimums
- Since consider only subsequences as motifs we introduce bias
- Note that Consensus can generate motif not seen in our data
19
A randomized approach to Motif finding¶

- One way to avoid bias and local minima is to introduce randomness
- We can generate candidate motifs from our data by treating it as distribution
- likely motif candidates from this distribution are those generated by Consensus
- Consensus strings can be tested by Scan-and-Score and their alignments lead to new consensus strings
- Eventually, we should converge to some local minimal answer
- To avoid finding a local minimum, we try several random starts, and search for the best score amongst all these starts.
- A randomized algorithm does not guarantee an optimal solution. Instead it promises a good/plausible answer on average, and it is not susceptible to a worse-case data sets as our greedy/biased method was.
20
Code for Randomized Motif Search¶
import random
def RandomizedMotifSearch(DNA,k):
""" Searches for a k-length motif that appears
in all given DNA sequences. It begins with a
random set of candidate consensus motifs
derived from the data. It refines the motif
until a true consensus emerges."""
# Seed motifs from random alignments
motifSet = set()
for i in xrange(500):
randomAlignment = [random.randint(0,len(DNA[j])-k) for j in xrange(len(DNA))]
motif = Consensus(randomAlignment, DNA, k)
motifSet.add(motif)
bestAlignment = []
minHammingDist = k*len(DNA)
kmer = ''
testSet = motifSet.copy()
while (len(testSet) > 0):
print len(motifSet),
nextSet = set()
for motif in testSet:
align, dist = ScanAndScoreMotif(DNA, motif)
# add new motifs based on these alignments
newMotif = Consensus(align, DNA, k)
if (newMotif not in motifSet):
nextSet.add(newMotif)
if (dist < minHammingDist):
bestAlignment = [s for s in align]
minHammingDist = dist
kmer = motif
testSet = nextSet.copy()
motifSet = motifSet | nextSet
return bestAlignment, minHammingDist, kmer
%time RandomizedMotifSearch(seqApprox,10)
Randomized algorithms should be restarted multiple times to insure a stable solution.
for i in xrange(10):
print RandomizedMotifSearch(seqApprox,10)
21
Lessons Learned¶
- We can find Motifs in our lifetime
- Practical exhaustive search algorithm for small k, MedianStringMotifSearch()
- Practical fast algorthim RandomizedMotifSearch(DNA,k)
- Three algorithm design approaches "Branch-and-Bound", "Greedy", and "Randomized"
- Reversing the objective, pretending that you know the answer, and validating it
- The power of randomness
- Not susceptable to worse case data
- Avoids local minimums that plague some greedy algorithms
22